REKLAMA
Dziennik Ustaw - rok 2018 poz. 1626
ROZPORZĄDZENIE
MINISTRA ZDROWIA1)
z dnia 16 sierpnia 2018 r.
zmieniające rozporządzenie w sprawie wymagań dotyczących oznakowania opakowań produktu leczniczego i treści ulotki2)
Na podstawie art. 26 ust. 2 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. z 2017 r. poz. 2211, z późn. zm.3)) zarządza się, co następuje:
§ 1. [Rozporządzenie w sprawie wymagań dotyczących oznakowania opakowań produktu leczniczego i treści ulotki]
1) w § 3 w ust. 1 po pkt 15 dodaje się pkt 15a w brzmieniu:
„15a) w przypadku produktu leczniczego, o którym mowa w art. 54a ust. 1 dyrektywy 2001/83/WE Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 r. w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi, zabezpieczenia, o których mowa w art. 54 lit. o tej dyrektywy lub art. 42a ust. 2 ustawy,”;
2) w § 6 w ust. 1:
a) w pkt 4 wprowadzenie do wyliczenia otrzymuje brzmienie:
„informacje niezbędne do prawidłowego stosowania produktu leczniczego, w szczególności:”,
b) w pkt 6 wprowadzenie do wyliczenia otrzymuje brzmienie:
„odniesienie do terminu ważności podanego na opakowaniu, wraz z następującymi informacjami:”,
c) pkt 7 otrzymuje brzmienie:
„7) nazwy produktu leczniczego w innych państwach członkowskich Unii Europejskiej, w przypadku gdy produkt został dopuszczony do obrotu w procedurze europejskiej, o ile są różne,”;
3) w § 7 w ust. 1 zdanie drugie otrzymuje brzmienie:
„Umieszczone symbole lub piktogramy muszą być zatwierdzone w procesie dopuszczenia do obrotu produktu leczniczego.”;
4) w § 14 w ust. 1 we wprowadzeniu do wyliczenia wyraz „wymienione” zastępuje się wyrazem „określone”;
5) § 18 otrzymuje brzmienie:
„§ 18. Treść ulotki i oznakowanie opakowań nie może zawierać elementów promocyjnych ani podawać właściwości czy wskazań do stosowania, które nie są zawarte w Charakterystyce Produktu Leczniczego, oraz nie może zachęcać do nieuzasadnionego wskazaniami stosowania produktu leczniczego.”;
6) w załączniku nr 1:
a) w ust. 1 „Nazwa produktu leczniczego” wyrazy „§ 3 ust. 1 pkt 1 lit. a” zastępuje się wyrazami „§ 3 ust. 1 pkt 1 lit. a–c”,
b) w ust. 2 „Skład jakościowy i ilościowy substancji czynnej” w części „Zawartość substancji czynnej” lit. C otrzymuje brzmienie:
„C. Różne dawki tej samej substancji określa się w ten sam sposób (niewłaściwe jest użycie w tekście np. 250 mg i 0,25 g), przy czym należy unikać ułamków dziesiętnych, gdyż przy pisaniu tych ułamków przecinek może być łatwo pominięty. Tak więc w zakresie od 1 do 999 mg dawkę należy podawać w miligramach, a nie w gramach. W celu uniknięcia pomyłki mikrogramy zapisuje się całym słowem, a nie skrótem jednostki, chyba że nie ma możliwości użycia czcionki wielkości nie mniejszej niż 7 punktów typograficznych, o których mowa w ust. 11. Dla produktów biologicznych, tam gdzie jest to możliwe, należy stosować jednostki międzynarodowe (j.m.).”,
c) w ust. 3 „Postać farmaceutyczna i jej zawartość w opakowaniu” akapit pierwszy otrzymuje brzmienie:
„Określając postać farmaceutyczną, należy korzystać z aktualnego ujednoliconego nazewnictwa Farmakopei Polskiej lub wykazu terminów standardowych Farmakopei Europejskiej. Lista ta zawiera również skrócone nazwy niektórych postaci farmaceutycznych, jednak można je stosować wyłącznie na małych etykietach, gdy nie ma miejsca na zamieszczenie pełnej nazwy drukowanej czcionką wielkości co najmniej 7 punktów typograficznych, o których mowa w ust. 11.”,
d) w ust. 5 lit. B otrzymuje brzmienie:
„B. Określenie terminu ważności po otwarciu opakowania
Jeżeli trwałość danego produktu leczniczego jest zmniejszona, np. w wyniku rozcieńczania, zmieszania z rozpuszczalnikiem lub otwarcia opakowania, podaje się maksymalny termin ważności produktu leczniczego. Ponadto, jeżeli maksymalny termin przydatności zależy od sposobu rozpuszczania lub rodzaju rozpuszczalnika, w informacji na opakowaniu zamieszcza się zalecenie zapoznania się z informacją znajdującą się w ulotce.
W przypadku niektórych produktów, takich jak produkty radiofarmaceutyczne lub niektóre szczepionki, może być konieczne dokładniejsze określenie zarówno terminu ważności, jak i czasu przechowywania po otwarciu pojemnika lub pierwszym użyciu produktu (podanie godziny, dnia, miesiąca i roku).
Na opakowaniach zamiast określenia „Terminu ważności” można użyć skrótu „EXP”, o ile dla określenia numeru serii użyto skrótu „Lot”, albo skrótu „Tw”, o ile dla określenia numeru serii użyto skrótu „S”, z koniecznością objaśnienia znaczenia skrótu na opakowaniu zewnętrznym lub bezpośrednim lub jeżeli to niemożliwe – w ulotce dla pacjenta w pkt 5: „Termin ważności (EXP)” albo „Termin ważności (Tw)”.”,
e) w ust. 6 „Warunki przechowywania” zdanie drugie otrzymuje brzmienie:
„Jeżeli to konieczne, zamieszcza się dalsze krótkie wyjaśnienie dotyczące warunków przechowywania, w sposób zrozumiały dla pacjenta.”,
f) ust. 9 otrzymuje brzmienie:
„9. Numer serii; dotyczy numeru zwalnianej serii u wytwórcy.
Na opakowaniach zamiast określenia „Numer serii” można użyć skrótu „Lot”, o ile dla określenia terminu ważności użyto skrótu „EXP”, albo skrótu „S”, o ile dla określenia terminu ważności użyto skrótu „Tw”, z koniecznością objaśnienia znaczenia skrótu na opakowaniu zewnętrznym lub bezpośrednim lub jeżeli to niemożliwe – w ulotce dla pacjenta w pkt 5: „Numer serii (Lot)” albo „Numer serii (S)”.”,
g) w ust. 10 pkt 1 i 2 otrzymują brzmienie:
„1) piktogram znaku drogowego zakazującego 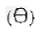
2) piktogram znaku drogowego ostrzegawczego 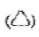
h) ust. 11 otrzymuje brzmienie:
„11. Do oznakowania opakowania produktu leczniczego stosuje się odpowiednio przepisy części II i III załącznika nr 2 do rozporządzenia. Informacje zamieszczane na oznakowaniu opakowań dla leków gotowych drukuje się czcionką wielkości co najmniej 7 punktów typograficznych (z zastrzeżeniem, że niezależnie od spełniania powyższego wymogu co do samej liczby punktów pozwala się drukować również czcionką wielkości takiej, aby wysokość „x” wynosiła co najmniej 1,4 mm), z odstępem między wierszami wynoszącym co najmniej 3 mm.”,
i) w ust. 12 zdanie pierwsze otrzymuje brzmienie:
„Jeżeli w przypadku niektórych produktów leczniczych spełnienie wymagań określonych w ust. 11 jest utrudnione, należy podejmować wszelkie możliwe działania w celu zwiększenia powierzchni oznakowania bez zmiany rozmiaru opakowania, np. etykietę można wydłużyć i poszerzyć albo tekst obrócić o 90° bez zmiany wielkości opakowania.”;
7) w załączniku nr 2 część I otrzymuje brzmienie:
„I. Rozmiar i rodzaj czcionki
Informacje zamieszczane w ulotkach leków gotowych drukuje się czcionką wielkości co najmniej 8 punktów typograficznych, z odstępem między wierszami wynoszącym co najmniej 3 mm.
Należy unikać pisania słów wielkimi literami (wersalikami). Rodzaj czcionki musi być łatwo czytelny.”.
§ 2. [Wejście w życie]
Minister Zdrowia: wz. J. Szczurek-Żelazko
1) Minister Zdrowia kieruje działem administracji rządowej - zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 10 stycznia 2018 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. poz. 95).
2) Niniejsze rozporządzenie w zakresie swojej regulacji:
1) wdraża dyrektywę Parlamentu Europejskiego i Rady 2011/62/UE z dnia 8 czerwca 2011 r. zmieniającą dyrektywę 2001/83/WE w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi - w zakresie zapobiegania wprowadzaniu sfałszowanych produktów leczniczych do legalnego łańcucha dystrybucji (Dz. Urz. UE L 174 z 01.07.2011, str. 74);
2) służy stosowaniu rozporządzenia delegowanego Komisji (UE) 2016/161 z dnia 2 października 2015 r. uzupełniającego dyrektywę 2001/83/WE Parlamentu Europejskiego i Rady przez określenie szczegółowych zasad dotyczących zabezpieczeń umieszczanych na opakowaniach produktów leczniczych stosowanych u ludzi (Dz. Urz. UE L 32 z 09.02.2016, str. 1).
3) Zmiany tekstu jednolitego wymienionej ustawy zostały ogłoszone w Dz. U. z 2018 r. poz. 650, 697, 1039, 1375, 1515 i 1544.
- Data ogłoszenia: 2018-08-23
- Data wejścia w życie: 2018-08-24
- Data obowiązywania: 2018-08-24

REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA