REKLAMA
Dziennik Ustaw - rok 2017 poz. 2051
ROZPORZĄDZENIE
MINISTRA ZDROWIA1)
z dnia 16 października 2017 r.
w sprawie leczenia krwią i jej składnikami w podmiotach leczniczych wykonujących działalność leczniczą w rodzaju stacjonarne i całodobowe świadczenia zdrowotne2)
Na podstawie art. 21 ust. 8 ustawy z dnia 22 sierpnia 1997 r. o publicznej służbie krwi (Dz. U. z 2017 r. poz. 1371) zarządza się, co następuje:
Rozdział 1
Przepisy ogólne
§ 1. [Zakres regulacji]
1) sposób i organizację leczenia krwią w podmiotach leczniczych wykonujących działalność leczniczą w rodzaju stacjonarne i całodobowe świadczenia zdrowotne, w których przebywają pacjenci ze wskazaniami do leczenia krwią i jej składnikami, zwanych dalej „podmiotami leczniczymi”, w tym zadania kierownika podmiotu leczniczego, ordynatora albo lekarza kierującego oddziałem oraz lekarzy, pielęgniarek i położnych;
2) organizację banku krwi oraz pracowni serologii lub pracowni immunologii transfuzjologicznej podmiotu leczniczego, a także sposób sprawowania nadzoru nad działaniem banku krwi oraz pracowni serologii lub pracowni immunologii transfuzjologicznej w tym podmiocie leczniczym, w tym wymagane kwalifikacje i doświadczenie kierownika banku krwi lub pracowni serologii lub pracowni immunologii transfuzjologicznej oraz jego zadania;
3) sposób prowadzenia dokumentacji medycznej dotyczącej leczenia krwią i jej składnikami;
4) sposób zapewnienia dostępu do badań z zakresu immunologii transfuzjologicznej.
2. Ilekroć w rozporządzeniu jest mowa o:
1) pracowni immunologii transfuzjologicznej – rozumie się przez to również pracownię serologii;
2) walidacji – rozumie się przez to przedstawienie obiektywnych dowodów potwierdzających, że zostały spełnione szczególne wymagania dotyczące zamierzonego, specyficznego zastosowania określonej procedury albo procesu.
Rozdział 2
Organizacja leczenia krwią i jej składnikami w jednostkach lub komórkach organizacyjnych podmiotu leczniczego
§ 2. [Organizacja leczenia krwią]
1) niezwłoczne, całodobowe zaopatrzenie jednostek lub komórek organizacyjnych zakładu leczniczego w krew i jej składniki;
2) badania z zakresu immunologii transfuzjologicznej, zwane dalej „badaniami immunohematologicznymi”, takie jak grupa krwi i próba zgodności krwi dawcy i biorcy przed przetoczeniem, zwana dalej „próbą zgodności”, warunkujące bezpieczne przetaczanie krwi i jej składników, zwane dalej „przetoczeniem”, oraz kwalifikujące kobiety RhD ujemne do podania immunoglobuliny anty-RhD podczas ciąży lub po porodzie;
3) identyfikację i rejestrowanie wszelkich niepożądanych zdarzeń i niepożądanych reakcji, w szczególności poważnych niepożądanych zdarzeń i poważnych niepożądanych reakcji związanych z przetoczeniem, a także sporządzanie raportów o tych zdarzeniach i reakcjach.
§ 3. [Odpowiedzialność lekarza]
1) ustalenie wskazań do przetoczenia;
2) identyfikację biorcy krwi na podstawie danych, o których mowa w § 11 ust. 1 pkt 1 i 2, i kontrolę dokumentacji medycznej przed przetoczeniem;
3) zabieg przetoczenia;
4) prawidłowe udokumentowanie zabiegu przetoczenia;
5) sporządzanie raportów o niepożądanych zdarzeniach i niepożądanych reakcjach, w tym poważnych niepożądanych zdarzeniach i reakcjach.
2. Pielęgniarka lub położna jest odpowiedzialna za:
1) czynności związane z pobieraniem próbek krwi od pacjentów;
2) identyfikację biorcy krwi, na podstawie danych, o których mowa w § 11 ust. 1 pkt 1 i 2, i kontrolę dokumentacji medycznej przed przetoczeniem;
3) obserwację biorcy krwi w trakcie przetoczenia i po przetoczeniu, niezwłoczne informowanie lekarza o objawach występujących w trakcie przetoczenia i po przetoczeniu mogących świadczyć o niepożądanych reakcjach, w tym poważnych niepożądanych reakcjach;
4) przekazywanie wypełnionego i podpisanego przez lekarza zlecenia na badania immunohematologiczne oraz badania kwalifikujące do podania immunoglobuliny anty-RhD oraz zamówienia na krew lub jej składniki do banku krwi lub właściwego centrum, o którym mowa w art. 23 ust. 3a, 4a i 5a ustawy z dnia 22 sierpnia 1997 r. o publicznej służbie krwi, zwanej dalej „ustawą”;
5) prawidłowe udokumentowanie zabiegu przetoczenia;
6) niezwłoczne informowanie lekarza o niepożądanych zdarzeniach, w tym poważnych niepożądanych zdarzeniach.
3. Naczelna pielęgniarka lub położna lub osoba nadzorująca pracę pielęgniarek lub położnych, w porozumieniu z ordynatorem lub inną osobą kierującą jednostką lub komórką organizacyjną zakładu leczniczego, ustala imienną listę pielęgniarek lub położnych uprawnionych do dokonywania przetoczeń i czynności związanych z tym zabiegiem, posiadających zaświadczenie o odbyciu szkolenia określone w przepisach wydanych na podstawie art. 21 ust. 7 ustawy i przedstawia do zatwierdzenia kierownikowi podmiotu leczniczego.
§ 4. [Zadania lekarzy, pielęgniarek i położnych wykonujących czynności związane z przetoczeniem]
1) wypełnienie zlecenia na badania immunohematologiczne oraz zamówienia na krew i jej składniki – dotyczy wyłącznie lekarza;
2) złożenie zamówienia na krew i jej składniki;
3) pobranie od pacjenta próbek krwi w celu wykonania badania grupy krwi i próby zgodności;
4) poinformowanie pacjenta o ryzyku i korzyściach wynikających z przetoczenia – dotyczy wyłącznie lekarza;
5) identyfikacja biorcy krwi na podstawie danych, o których mowa w § 11 ust. 1 pkt 1 i 2, i kontrola dokumentacji medycznej przed zabiegiem przetoczenia;
6) zabieg przetoczenia;
7) obserwacja pacjenta w trakcie przetoczenia i po przetoczeniu oraz podjęcie odpowiednich czynności, jeżeli wystąpi niepożądana reakcja.
2. Informacje o zabiegu przetoczenia i niepożądanych zdarzeniach i niepożądanych reakcjach, w tym poważnych niepożądanych zdarzeniach i poważnych niepożądanych reakcjach, mających miejsce w trakcie przetoczenia lub bezpośrednio po przetoczeniu, odnotowuje się w historii choroby, książce transfuzyjnej, której wzór jest określony w załączniku nr 1 do rozporządzenia, karcie informacyjnej z leczenia szpitalnego pacjenta oraz księdze raportów pielęgniarskich.
3. Wpisu w książce transfuzyjnej dokonuje pielęgniarka lub położna, przy czym każdy wpis musi być sprawdzony przez lekarza odpowiedzialnego za przetoczenie i potwierdzony. Potwierdzenie obejmuje wskazanie imienia, nazwiska lekarza, tytułu zawodowego, specjalizacji, numeru prawa wykonywania zawodu oraz czytelny podpis.
4. Dokumentacja wskazana w ust. 2 może być prowadzona w postaci elektronicznej w sposób określony w przepisach wydanych na podstawie art. 30 ust. 1 ustawy z dnia 6 listopada 2008 r. o prawach pacjenta i Rzeczniku Praw Pacjenta (Dz. U. z 2017 r. poz. 1318 i 1524).
§ 5. [Lekarz odpowiedzialny za gospodarkę krwią]
2. W przypadku gdy w podmiocie leczniczym nie zatrudnia się lekarza specjalisty w dziedzinie transfuzjologii klinicznej, obowiązki lekarza odpowiedzialnego za gospodarkę krwią powierza się innemu lekarzowi specjaliście, który wykonuje czynności w jednostkach lub komórkach organizacyjnych zakładu leczniczego, w których często przetacza się krew i jej składniki.
3. Lekarz odpowiedzialny za gospodarkę krwią:
1) odbywa przeszkolenie w jednej z jednostek organizacyjnych publicznej służby krwi, o których mowa w art. 4 ust. 3 ustawy, nie rzadziej niż co 4 lata;
2) bierze udział w kursach i seminariach organizowanych przez jednostki organizacyjne publicznej służby krwi, o których mowa w art. 4 ust. 3 ustawy.
4. Do zadań lekarza odpowiedzialnego za gospodarkę krwią należy:
1) nadzór nad leczeniem krwią i jej składnikami w jednostkach lub komórkach organizacyjnych zakładu leczniczego;
2) planowanie zaopatrzenia podmiotu leczniczego w krew i jej składniki;
3) kierowanie bankiem krwi, jeżeli tej czynności nie powierzono kierownikowi pracowni immunologii transfuzjologicznej;
4) nadzorowanie przestrzegania SOP przez bank krwi, pracownię immunologii transfuzjologicznej lub inne jednostki lub komórki organizacyjne zakładu leczniczego;
5) organizacja wewnętrznych szkoleń lekarzy i pielęgniarek lub położnych w dziedzinie leczenia krwią w podmiocie leczniczym oraz szkoleń dla pracowników banku krwi, w zakresie zadań określonych w § 16 ust. 2;
6) przekazywanie do instytutu, o którym mowa w art. 4 ust. 3 pkt 1 ustawy, za pośrednictwem właściwego centrum raportów o niepożądanych zdarzeniach i niepożądanych reakcjach;
7) sporządzanie i przekazywanie do właściwego centrum rocznych sprawozdań z działalności podmiotu leczniczego w zakresie krwiolecznictwa, nie później niż do dnia 30 stycznia każdego roku, za rok poprzedni.
§ 6. [Standardowa procedura operacyjna (SOP)]
2. Zmiany w wykonywaniu danej procedury wprowadza się, sporządzając nową SOP lub uaktualniając dotychczasową wersję SOP.
3. Czynności określone w rozdziałach 2–5 opisuje się w SOP.
4. Kierownik podmiotu leczniczego lub wyznaczona przez niego osoba przechowuje oryginał SOP.
5. Kopie SOP sporządza się dla każdego stanowiska pracy związanego z leczeniem krwią i jej składnikami.
6. Kopie SOP, o których mowa w ust. 5, przechowuje się przy stanowisku pracy i u kierownika jednostki lub komórki organizacyjnej zakładu leczniczego.
7. SOP sporządza się według wzoru określonego w załączniku nr 2 do rozporządzenia.
8. SOP zatwierdza kierownik podmiotu leczniczego.
9. Oryginały nieaktualnych SOP należy przechowywać 30 lat.
§ 7. [Wpis dokonany w dokumentacji dotyczący leczenia krwią]
2. Poprawek lub zmian w dokumentacji należy dokonywać w taki sposób, aby możliwe było jednoznaczne rozróżnienie danych pierwotnych od danych, które poprawiono lub zmieniono.
3. Poprawkę lub zmianę należy każdorazowo opatrzyć podpisem osoby, która tę poprawkę lub zmianę wprowadziła, oraz datą jej dokonania.
§ 8. [Komitet transfuzjologiczny]
1) rozwiązywania problemów dotyczących leczenia krwią i jej składnikami;
2) rozwiązywania problemów związanych z gospodarką krwią i jej składnikami;
3) sprawowania nadzoru nad leczeniem krwią i jej składnikami.
2. W skład komitetu wchodzą:
1) ordynatorzy lub inne osoby kierujące jednostkami lub komórkami organizacyjnymi zakładu leczniczego, w których często przetacza się krew i jej składniki, lub ich zastępcy;
2) lekarz odpowiedzialny za gospodarkę krwią;
3) lekarz specjalista anestezjologii i intensywnej terapii;
4) kierownik pracowni immunologii transfuzjologicznej;
5) pielęgniarka lub położna dokonująca przetoczeń.
3. Komitet współpracuje z jednostkami organizacyjnymi publicznej służby krwi, o których mowa w art. 4 ust. 3 ustawy, w szczególności z właściwym centrum.
4. Do zadań komitetu należy w szczególności:
1) dokonywanie okresowej oceny wskazań do przetoczenia, nie rzadziej niż raz na 6 miesięcy;
2) analiza zużycia krwi i jej składników w celu ograniczenia niepotrzebnych przetoczeń i nadmiernych zniszczeń krwi i jej składników;
3) nadzór nad działaniami związanymi z leczeniem krwią lub jej składnikami oraz nadzór nad związaną z tym dokumentacją;
4) ocena prawidłowości postępowania podczas przetoczeń wykonanych w podmiocie leczniczym, nie rzadziej niż raz na 6 miesięcy;
5) analiza każdego niepożądanego zdarzenia i każdej niepożądanej reakcji wraz z oceną postępowania;
6) analiza raportów o niepożądanych zdarzeniach i reakcjach;
7) opracowywanie wewnętrznych programów kształcenia lekarzy i pielęgniarek lub położnych w dziedzinie leczenia krwią i jej składnikami oraz nadzór nad ich realizacją;
8) udział w planowaniu zaopatrzenia w krew i jej składniki oraz w rocznej sprawozdawczości dotyczącej ich zużycia.
5. Raporty i okresowe sprawozdania z działalności komitetu są przekazywane kierownikowi podmiotu leczniczego i kierownikowi właściwego centrum nie rzadziej niż raz na rok, najpóźniej do dnia 30 stycznia każdego roku, za rok poprzedni.
6. Specjalistyczny nadzór nad leczeniem krwią i jej składnikami, o którym mowa w art. 27 ust. 1 pkt 12 ustawy, sprawuje właściwe centrum; w ramach sprawowanego nadzoru właściwe centrum przeprowadza kontrolę, o której mowa w art. 29 ust. 1 ustawy, co najmniej raz na dwa lata.
§ 9. [Wypis zamówienia indywidualnego na krew lub jej składniki]
2. Wiarygodnym wynikiem będącym podstawą wypisania zamówienia, o którym mowa w ust. 1, może być wyłącznie wynik grupy krwi oparty na dwóch oznaczeniach wykonanych z dwóch próbek krwi pobranych od tego samego pacjenta w różnym czasie:
1) wpisany w karcie identyfikacyjnej grupy krwi, której wzór jest określony w załączniku nr 4 do rozporządzenia (karta jest ważna tylko łącznie z dokumentem tożsamości), albo
2) wpisany w legitymacji służbowej żołnierzy zawodowych, albo
3) pochodzący z pracowni immunologii transfuzjologicznej zawierający wpisy o dwóch oznaczeniach wykonanych w różnym czasie; jeżeli wynik pochodzi tylko z badania jednej próbki, lekarz zleca wykonanie drugiego oznaczania grupy krwi przed wydaniem składnika krwi; w przypadku koncentratu krwinek czerwonych, zwanego dalej „KKCz”, lub krwi pełnej konserwowanej, zwanej dalej „KPK”, badanie to może być wykonane przy próbie zgodności.
3. W przypadku braku wyniku z dwukrotnego oznaczenia grupy krwi lub jeżeli istnieją wątpliwości co do wiarygodności wyniku badania grupy krwi, lekarz zleca wykonanie kolejnego oznaczenia grupy krwi przed wydaniem składnika krwi; w przypadku KKCz lub KPK oznaczenie grupy krwi może być wykonane przy próbie zgodności.
4. W przypadku braku wiarygodnego wyniku, jeżeli przetoczenie jest pilne, lekarz postępuje zgodnie z przepisami ust. 10 i 11.
5. Próbkę krwi pobiera się na podstawie wypełnionego i podpisanego przez lekarza zlecenia na badanie grupy krwi, którego wzór jest określony w załączniku nr 5 do rozporządzenia, lub zlecenia na wykonanie próby zgodności, którego wzór jest określony w załączniku nr 6 do rozporządzenia.
6. Na podstawie wyniku badania grupy krwi, którego wzór jest określony w załączniku nr 7 do rozporządzenia, lekarz wypełnia zamówienie indywidualne na krew i jej składniki, które przekazuje się do banku krwi.
7. Jeżeli w podmiocie leczniczym nie ma banku krwi, zamówienie indywidualne na krew i jej składniki przekazuje się bezpośrednio do właściwego centrum.
8. W przypadku planowanego przetoczenia KKCz, KPK lub koncentratu granulocytarnego, zwanego dalej „KG”, do banku krwi przekazuje się zamówienie indywidualne na krew lub jej składniki oraz zlecenie wykonania próby zgodności, wraz z odrębnie w tym celu pobraną próbką krwi od pacjenta.
9. Przed wydaniem z banku krwi próbek krwi dawców do badań immunohematologicznych pracownik sprawdza zgodność grupy krwi i numeru donacji na segmencie drenu, będącego próbką krwi dawcy, z grupą krwi i numerem donacji na etykiecie pojemnika i po wpisaniu na zleceniu tych danych przekazuje zlecenie wraz z próbką krwi, o której mowa w ust. 5, do pracowni immunologii transfuzjologicznej.
10. W przypadku bezpośredniego zagrożenia życia lekarz może podjąć decyzję o przetoczeniu KKCz albo KPK zgodnych w układzie ABO i RhD przed wykonaniem próby zgodności na podstawie wiarygodnego wyniku grupy krwi określonego w ust. 2. W innych przypadkach lekarz podejmuje decyzję o przetoczeniu osocza grupy AB, koncentratu krwinek płytkowych, zwanego dalej „KKP”, rekonstytuowanego grupy O zawieszonego w osoczu AB lub w roztworze wzbogacającym lub KKP grupy AB oraz KKCz grupy O, a w przypadku pacjentów z alloprzeciwciałami anty-D, dziewczynek oraz kobiet w wieku rozrodczym, KKCz grupy O RhD ujemny, K ujemny, jeżeli u pacjentki nie wykryto lub nie badano antygenu K. Przy braku KKCz O RhD ujemnego, K ujemnego dopuszcza się przetoczenie KKCz grupy O RhD dodatni.
11. W przypadku, o którym mowa w ust. 10, do banku krwi przekazuje się zamówienie na krew lub jej składniki do pilnego przetoczenia, którego wzór jest określony w załączniku nr 8 do rozporządzenia. Następnie lekarz wypełnia zlecenie na badanie grupy krwi ABO i RhD, jeżeli brak jest wyniku, oraz wypełnia zlecenie wykonania próby zgodności. Dalsze postępowanie prowadzi się zgodnie z wymogami określonymi w obwieszczeniu wydanym na podstawie art. 24 pkt 2 lit. b ustawy.
§ 10. [Pobieranie krwi]
2. Bezpośrednio przed pobraniem osoba pobierająca dokonuje jednoznacznej identyfikacji i weryfikacji tożsamości pacjenta. Na etykiecie probówki, w obecności pacjenta, na podstawie danych uzyskanych od pacjenta, a jeżeli jest to niemożliwe – danych uzyskanych na podstawie stosowanego w podmiocie leczniczym znaku identyfikacyjnego, wpisuje się następujące dane:
1) nazwisko i imię pacjenta (wielkimi literami);
2) numer PESEL pacjenta, a w przypadku braku numeru PESEL – datę urodzenia pacjenta;
3) datę i godzinę pobrania próbki krwi.
3. W przypadku braku możliwości uzyskania danych pacjenta, o których mowa w ust. 2, na etykiecie i na zleceniu na badanie grupy krwi należy wpisać symbol „NN”, płeć, numer księgi głównej lub niepowtarzalny numer identyfikacyjny pacjenta, o którym mowa w przepisach wydanych na podstawie art. 36 ust. 6 ustawy z dnia 15 kwietnia 2011 r. o działalności leczniczej (Dz. U. z 2016 r. poz. 1638, 1948 i 2260).
4. Po pobraniu próbki krwi i opisaniu probówki osoba pobierająca sprawdza, czy dane pacjenta na etykiecie probówki z próbką krwi pacjenta są zgodne z danymi na zleceniu na badanie grupy krwi lub na zleceniu na wykonanie próby zgodności, i składa na tych zleceniach czytelny podpis oraz wpisuje datę i godzinę pobrania.
5. Pobrana próbka krwi jest niezwłocznie dostarczana do pracowni immunologii transfuzjologicznej wraz ze zleceniem na badanie grupy krwi lub zleceniem na wykonanie próby zgodności oraz zleceniem badania kwalifikacyjnego do podania immunoglobuliny anty-RhD w przypadku kobiet RhD ujemnych.
6. W przypadku korzystania z systemu teleinformatycznego jest drukowana etykieta z danymi, o których mowa w ust. 2, a zlecenie, o którym mowa w ust. 4, jest przekazywane w postaci elektronicznej, z zastosowaniem podpisu elektronicznego, w sposób określony w przepisach wydanych na podstawie art. 30 ust. 1 ustawy z dnia 6 listopada 2008 r. o prawach pacjenta i Rzeczniku Praw Pacjenta.
§ 11. [Kontrola zgodności grupy krwi biorcy]
1) identyfikacji pacjenta oraz porównaniu jego imienia i nazwiska, numeru PESEL lub daty urodzenia z danymi zawartymi w wyniku próby zgodności, którego wzór jest określony w załączniku nr 9 do rozporządzenia;
2) identyfikacji pacjenta opisanego symbolem NN, symbolem płci oraz porównanie przypisanego numeru księgi głównej lub niepowtarzalnego numeru identyfikacyjnego pacjenta z danymi zawartymi w wyniku grupy krwi lub wyniku próby zgodności;
3) porównaniu wyniku badania grupy krwi pacjenta z grupą krwi na etykiecie pojemnika oraz, w przypadku przetaczania KKCz, KPK i KG, dodatkowo z grupą krwi zawartą w wyniku próby zgodności;
4) porównaniu numeru donacji krwi lub jej składnika z numerem donacji zawartym w wyniku próby zgodności, jeżeli jest wymagany;
5) sprawdzeniu, czy jednostka krwi lub jej składnika została przygotowana zgodnie ze specjalnymi zaleceniami wpisanymi w karcie zleceń lekarskich;
6) sprawdzeniu daty ważności krwi lub jej składnika;
7) sprawdzeniu daty ważności próby zgodności zawartej w wyniku próby zgodności;
8) porównaniu wyniku grupy krwi pacjenta z grupą krwi na etykiecie składnika, w przypadku przetaczania KKP, osocza lub krioprecypitatu.
2. Lekarz i uprawniona do tego pielęgniarka lub położna, którzy dokonali oceny zgodności krwi lub jej składnika z grupą krwi biorcy krwi, składają swój podpis na wyniku próby zgodności wraz z datą i godziną dokonania oceny lub na innym dokumencie wydanym przez bank krwi jednoznacznie wskazującym, dla kogo dany składnik krwi jest przeznaczony.
3. W przypadku przetaczania składnika krwi niewymagającego przed podaniem wykonania próby zgodności: osocza, KKP oraz krioprecypitatu, lekarz i uprawniona do tego pielęgniarka albo położna składają swój podpis na druku rozchodu, który jest wydawany ze składnikiem krwi, z datą i godziną oraz umieszczają adnotację dokonania oceny prawidłowości doboru składnika w historii choroby pacjenta.
4. Datę i godzinę rozpoczęcia przetoczenia zawartości każdego pojemnika należy wpisać w książce transfuzyjnej, w wyniku próby zgodności oraz w karcie znieczulenia ogólnego, jeżeli obowiązuje, a na oddziale anestezjologii i intensywnej terapii – w karcie obserwacji pacjenta, oraz w dokonanej ocenie zgodności z wynikiem grupy krwi pacjenta.
5. W przypadku rozbieżności wykrytych podczas kontroli zgodności krwi lub jej składnika z danymi biorcy krwi, nie wolno przetaczać tej jednostki krwi lub tego składnika.
6. W przypadku, o którym mowa w ust. 5, krew lub jej składnik zwraca się do banku krwi wraz z protokołem zawierającym informację o przyczynie zwrotu i wynikiem próby zgodności. Krew lub jej składniki nie mogą być ponownie dopuszczone do użytku.
7. W przypadku, o którym mowa w ust. 5, sporządza się raport o niepożądanym zdarzeniu.
8. Wynik próby zgodności, jeżeli jest wymagany, oraz wynik grupy krwi muszą być dostępne podczas przetoczenia.
§ 12. [Zasady dotyczące przetaczania]
2. Przetoczenie KKCz, KPK i KG należy rozpocząć nie później niż w okresie 30 minut od ich wydania z banku krwi.
3. Pojedyncze jednostki krwi lub jej składników należy sukcesywnie pobierać z banku krwi.
4. W wyjątkowych przypadkach można wydawać jednorazowo więcej niż jedną jednostkę krwi lub jej składników, pod warunkiem że będą one przechowywane w oddziale w warunkach wymaganych dla danego składnika krwi. Proces przechowywania musi być poddawany systematycznej walidacji oraz kontroli bieżącej.
5. Jeżeli przewiduje się, że czas do rozpoczęcia przetoczenia KKCz lub KPK będzie dłuższy niż 30 minut od wydania z banku krwi, należy je przechowywać w chłodziarce przeznaczonej wyłącznie do tego celu, w której proces przechowywania został poddany walidacji, w temperaturze od 2°C do 6°C, przy czym temperaturę w chłodziarce należy sprawdzać i zapisywać co najmniej 3 razy w ciągu doby (co 8 godzin).
6. Planowane przetoczenia powinny odbywać się w okresie pełnej obsady lekarzy i pielęgniarek lub położnych jednostki lub komórki organizacyjnej zakładu leczniczego.
7. Składniki krwi przetacza się za pomocą jednorazowych sterylnych zestawów.
8. Nie można przetaczać przez zestaw uprzednio użyty do przetaczania KPK lub KKCz jakichkolwiek składników krwi i płynów infuzyjnych, z zastrzeżeniem ust. 15 i 16.
9. Do przetoczeń niemowlętom służą specjalne zestawy. Jeżeli składnik krwi jest podawany strzykawką, należy zastosować specjalny filtr.
10. Używane pompy muszą posiadać oznakowanie CE i instrukcję wytwórcy, jak należy je stosować.
11. Ogrzewanie KKCz lub KPK można przeprowadzać wyłącznie w specjalistycznym urządzeniu zaopatrzonym w termometr i system alarmowy. Zaleca się ich ogrzewanie, do temperatury nie wyższej niż 37°C, w przypadku:
1) dorosłych – jeżeli szybkość przetoczenia przekracza 50 ml/min;
2) dzieci – jeżeli szybkość przetoczenia przekracza 15 ml/min;
3) noworodków – w przetoczeniu wymiennym;
4) biorców z klinicznie znaczącymi przeciwciałami typu zimnego.
12. Nie można dodawać produktów leczniczych do przetaczanej krwi lub jej składników.
13. Nie można przetaczać jednej jednostki krwi pełnej lub KKCz dłużej niż 4 godziny, a jednej jednostki KKP, osocza albo krioprecypitatu – dłużej niż 30 minut.
14. Nie można po odłączeniu ponownie podłączać biorcy krwi tego samego zestawu lub tego samego pojemnika ze składnikiem krwi.
15. Przez jeden zestaw można przetaczać, podczas jednego zabiegu, jedno opakowanie krwi lub jej składnika. Zestaw należy zmienić również w przypadku, gdy po zakończonym przetoczeniu podaje się płyny infuzyjne. W przypadkach wymagających szybkiego przetaczania kilku albo kilkunastu jednostek KKCz, KKP, KPK oraz osocza (masywna transfuzja), dopuszcza się przetaczanie z użyciem jednego zestawu do przetaczania, pod warunkiem, że przetoczenie będzie przeprowadzone przy użyciu specjalistycznego sprzętu.
16. Dopuszcza się przetaczanie podczas jednego zabiegu przetoczenia kilku jednostek krioprecypitatu przez jeden zestaw do przetaczania.
17. Krew lub jej składniki niewykorzystane w całości nie mogą być przetoczone innemu pacjentowi.
18. Pojemniki z pozostałością składnika krwi po przetoczeniu wraz z zestawami do przetoczenia, opisane nazwiskiem i imieniem pacjenta oraz datą i godziną przetoczenia, należy przechowywać w temperaturze od 2°C do 6°C przez 72 godziny w specjalnie do tego celu przeznaczonej chłodziarce, a następnie zutylizować w sposób uniemożliwiający pozyskanie danych osobowych pacjenta przez osoby nieuprawnione.
19. Pojemniki, o których mowa w ust. 18, muszą być odpowiednio zabezpieczone przed rozlaniem i wtórnym zakażeniem, chłodziarki przeznaczone do ich przechowywania muszą podlegać wstępnej i okresowej kwalifikacji, a proces przechowywania – systematycznej walidacji. Pomiar temperatury w chłodziarce powinien być przeprowadzany co najmniej 3 razy w ciągu doby (co 8 godzin) i dokumentowany.
§ 13. [Odpowiedzialność lekarzy, pielęgniarek i położnych]
2. Bezpośrednio przed przetoczeniem, po 15 minutach od rozpoczęcia przetoczenia każdej jednostki krwi lub jej składnika oraz po jego zakończeniu, należy zmierzyć i zarejestrować ciepłotę ciała, tętno i ciśnienie tętnicze krwi pacjenta.
3. Lekarz odpowiedzialny za przetoczenie lub wyznaczona przez niego pielęgniarka lub położna oraz lekarz przejmujący opiekę nad pacjentem są odpowiedzialni za obserwację pacjenta podczas przetoczenia i przez 12 godzin po jego zakończeniu.
4. Pacjentowi należy udzielić informacji o konieczności niezwłocznego zgłoszenia każdego niepokojącego objawu, w szczególności dreszczy, wysypki, zaczerwienienia skóry, duszności oraz bólu kończyn lub okolicy lędźwiowej.
5. Lekarz lub pielęgniarka zwracają szczególną uwagę na pacjentów, którzy są nieprzytomni. Pogorszenie stanu ogólnego pacjenta, w szczególności w okresie od 15 do 20 minut od rozpoczęcia przetaczania każdej jednostki krwi lub jej składnika, może być objawem niepożądanej reakcji, o której świadczą: obniżenie ciśnienia tętniczego, nieuzasadnione krwawienie, które może być następstwem rozsianego wykrzepiania wewnątrznaczyniowego, oraz hemoglobinuria lub oliguria, które mogą być pierwszym objawem ostrej hemolitycznej reakcji poprzetoczeniowej.
§ 14. [Poważne niepożądane zdarzenia i poważne niepożądane reakcje]
1) reakcję hemolityczną;
2) zakażenie bakteryjne;
3) reakcję alergiczną lub anafilaktyczną;
4) ostre poprzetoczeniowe uszkodzenie płuc, zwane dalej „TRALI”;
5) duszność poprzetoczeniową;
6) niehemolityczną reakcję gorączkową;
7) poprzetoczeniowe przeciążenie krążenia (TACO).
2. Do opóźnionych niepożądanych reakcji zalicza się w szczególności:
1) reakcję hemolityczną;
2) poprzetoczeniową skazę małopłytkową;
3) poprzetoczeniową chorobę przeszczep przeciw biorcy (TA-GvHD);
4) przeniesienie biologicznych czynników chorobotwórczych.
3. W przypadku wystąpienia u pacjenta objawów nasuwających podejrzenie wczesnej niepożądanej reakcji, w tym poważnej niepożądanej reakcji, należy:
1) niezwłocznie przerwać przetoczenie i powiadomić lekarza;
2) odłączyć pojemnik ze składnikiem krwi wraz z zestawem do przetoczenia, utrzymując jednocześnie wkłucie do żyły, i powoli przetaczać choremu – przez nowy sterylny zestaw – 0,9% roztwór chlorku sodowego (NaCl) do czasu wdrożenia odpowiedniego leczenia;
3) zabezpieczyć odłączony składnik krwi do ewentualnych dalszych badań;
4) zmierzyć pacjentowi ciepłotę ciała, tętno i ciśnienie tętnicze krwi;
5) dalsze postępowanie uzależniać od nasilenia i rodzaju objawów.
4. W przypadku gdy potwierdzi się podejrzenie, że objawy wskazują na wystąpienie poważnej niepożądanej reakcji, należy niezwłocznie:
1) sprawdzić:
a) dane na wszystkich pojemnikach z przetaczaną krwią lub jej składnikami,
b) wynik próby zgodności i wynik grupy krwi pacjenta,
c) dane identyfikujące pacjenta, o których mowa w § 11 ust. 1 pkt 1 i 2;
2) pobrać próbki krwi od pacjenta z innego miejsca wkłucia niż miejsce, w którym dokonywano przetoczenia, w celu wykonania badań:
a) immunohematologicznych w zakresie ustalonym z pracownią badań konsultacyjnych centrum, a w przypadku podejrzenia TRALI – w zakresie ustalonym przez jednostkę organizacyjną publicznej służby krwi, o której mowa w art. 4 ust. 3 pkt 1 ustawy,
b) bakteriologicznych (na posiew); objętość próbki krwi i rodzaj pojemnika z podłożem bakteriologicznym określa pracownia mikrobiologiczna wykonująca badania dla podmiotu leczniczego;
3) powiadomić pracownię immunologii transfuzjologicznej, która wykonywała badania przed przetoczeniem; pracownia kontroluje dokumentację i ponownie wykonuje badania grupy krwi biorcy krwi i krwi dobranej do przetoczenia, a wyniki badań przekazuje do lekarza odpowiedzialnego za przetoczenie;
4) powiadomić właściwe centrum, pod którego nadzór specjalistyczny podlega terytorialnie dany podmiot leczniczy, w celu dalszego przeprowadzania badań; właściwe centrum dla podmiotu leczniczego powiadamia centrum, z którego otrzymano składniki krwi, o wystąpieniu poważnej niepożądanej reakcji lub poważnego niepożądanego zdarzenia;
5) przesłać do działu lub pracowni immunologii transfuzjologicznej właściwego centrum:
a) wszystkie próbki krwi pacjenta oraz krwi dobieranej do przetoczenia znajdujące się w pracowni immunologii transfuzjologicznej,
b) próbki krwi pacjenta pobrane do badań immunohematologicznych po przetoczeniu,
c) w przypadku podejrzenia TRALI – próbki krwi wraz ze zgłoszeniem niepożądanej reakcji lub zdarzenia, którego wzór jest określony w załączniku nr 10 do rozporządzenia; dział lub pracownia immunologii transfuzjologicznej właściwego centrum przesyła próbki do diagnostyki w jednostce organizacyjnej publicznej służby krwi, o której mowa w art. 4 ust. 3 pkt 1 ustawy;
6) przesłać do pracowni mikrobiologicznej uzgodnionej z właściwym centrum:
a) pobrane po przetoczeniu próbki krwi pacjenta,
b) wszystkie pojemniki z resztkami przetaczanej krwi lub jej składników; pracownia mikrobiologiczna po pobraniu z pojemników próbek krwi do badań przesyła je do działu lub pracowni immunologii transfuzjologicznej właściwego centrum;
7) w przypadku wystąpienia duszności poprzetoczeniowej – przeprowadzić badania w celu diagnostyki TRALI: badanie radiologiczne klatki piersiowej oraz badanie CRP (białko C-reaktywne) w surowicy;
8) przesłać do właściwego centrum zgłoszenie niepożądanej reakcji wypełnione przez lekarza odpowiedzialnego za przetoczenie.
5. W przypadku gdy niepożądana reakcja wystąpi po zakończonym przetoczeniu, należy powiadomić lekarza odpowiedzialnego za przetoczenie i postępować zgodnie z przepisami ust. 4 pkt 4–8.
6. W przypadkach wystąpienia poważnych niepożądanych zdarzeń i poważnych niepożądanych reakcji, kierownik właściwego centrum lub upoważniony przez niego lekarz przeprowadza w podmiocie leczniczym kontrolę postępowania przed przetoczeniem i podczas jego przeprowadzania oraz udziela wskazówek dotyczących postępowania po ich wystąpieniu.
Rozdział 3
Organizacja banku krwi w podmiocie leczniczym
§ 15. [Utworzenie banku krwi]
2. Kierownikiem banku krwi jest lekarz odpowiedzialny za gospodarkę krwią lub kierownik pracowni immunologii transfuzjologicznej.
§ 16. [Zadania banku krwi]
2. Do zadań banku krwi należy w szczególności:
1) składanie zamówień na krew i jej składniki we właściwym centrum, zgodnie z zamówieniami jednostek lub komórek organizacyjnych zakładu leczniczego;
2) odbiór krwi i jej składników;
3) przechowywanie krwi i jej składników do czasu ich wydania do jednostki lub komórki organizacyjnej zakładu leczniczego;
4) wydawanie krwi i jej składników do jednostek lub komórek organizacyjnych zakładu leczniczego;
5) prowadzenie dokumentacji dotyczącej przychodów i rozchodów krwi i jej składników;
6) sporządzanie sprawozdań dotyczących zużycia krwi i jej składników i przekazywanie ich do właściwego centrum;
3. Pracownicy banku krwi gromadzą i przekazują okresowo (co 3 miesiące) do właściwego centrum dane dotyczące niepożądanych reakcji poprzetoczeniowych i niepożądanych zdarzeń, które nie zostały zakwalifikowane jako poważne.
4. Kierownik banku krwi sporządza SOP obowiązujące w banku krwi, które zatwierdza kierownik podmiotu leczniczego. Warunkiem wykonywania działalności przez bank krwi jest posiadanie zatwierdzonych SOP.
§ 17. [Osoby wykonujące zadania banku krwi]
2. Nadzór nad czynnościami osób, o których mowa w ust. 1, sprawuje kierownik banku krwi.
§ 18. [Dokumentacja działalności banku krwi]
2. Książka przychodów i rozchodów zawiera w szczególności następujące informacje:
1) datę i godzinę przychodu;
2) nazwę, numer donacji, grupę krwi, ilość krwi lub jej składników, datę pobrania oraz datę ważności;
3) czytelny podpis zawierający imię i nazwisko osoby odbierającej;
4) datę i godzinę rozchodu;
5) nazwę jednostki lub komórki organizacyjnej zakładu leczniczego, do którego przekazano krew lub jej składniki;
6) imię, nazwisko, numer PESEL lub datę urodzenia pacjenta – w przypadku braku numeru PESEL; w przypadku braku danych pacjenta – symbol „NN”, płeć oraz numer księgi głównej lub niepowtarzalny numer identyfikacyjny pacjenta, o którym mowa w przepisach wydanych na podstawie art. 36 ust. 6 ustawy z dnia 15 kwietnia 2011 r. o działalności leczniczej;
7) czytelny podpis zawierający imię i nazwisko osoby wydającej krew lub jej składniki.
3. Wyniki kontroli temperatury w chłodziarkach, zamrażarkach i w innym sprzęcie do termostatowania, przeznaczonych do przechowywania krwi i jej składników, dokumentuje się przez sporządzenie protokołu kontroli temperatury przechowywania krwi i jej składników.
4. Protokoły kontroli temperatury przechowywania krwi i jej składników oraz protokoły kontroli temperatury transportu krwi i jej składników należy przechowywać przez okres co najmniej 5 lat od końca roku, w którym dokonano pomiarów.
5. Książka przychodów i rozchodów może być prowadzona w postaci elektronicznej w sposób określony w przepisach wydanych na podstawie art. 30 ust. 1 ustawy z dnia 6 listopada 2008 r. o prawach pacjenta i Rzeczniku Praw Pacjenta.
§ 19. [Zamówienie krwi i jej składników]
2. Składając wstępne zamówienie telefoniczne, należy uzgodnić z właściwym centrum termin i sposób dostarczenia krwi lub jej składników do banku krwi albo ich odbioru.
3. Uzupełniając zapas krwi i jej składników, bank krwi jest obowiązany sporządzić pisemne zamówienie zbiorcze na krew i jej składniki, którego wzór jest określony w załączniku nr 11 do rozporządzenia, i uzyskać jego akceptację przez kierownika podmiotu leczniczego lub osobę przez niego upoważnioną oraz głównego księgowego. Akceptacja taka może mieć formę stałej akceptacji rocznej.
4. Zamówienia indywidualne oraz zamówienia zbiorcze na krew i jej składniki należy dostarczyć do właściwego centrum przed wydaniem odbiorcy krwi lub jej składników.
5. W przypadkach pilnego przetoczenia krew i jej składniki można wydawać na podstawie zamówienia telefonicznego, zamówienia przesłanego faksem lub zamówienia przesłanego pocztą elektroniczną, z zachowaniem bezpieczeństwa danych. Zamówienie to musi być niezwłocznie uzupełnione formalnym złożeniem zamówienia na krew i jej składniki, najpóźniej w terminie 24 godzin od odbioru krwi lub jej składników.
6. Zamówienia indywidualne oraz zamówienia zbiorcze na krew i jej składniki sporządza się w dwóch egzemplarzach. Oryginał zamówienia przesyła się do właściwego centrum, a kopię przechowuje się w banku krwi przez okresy określone w § 18 ust. 1.
7. Zamówienie zbiorcze i zamówienie indywidualne mogą być prowadzone w postaci elektronicznej w sposób określony w przepisach wydanych na podstawie art. 30 ust. 1 ustawy z dnia 6 listopada 2008 r. o prawach pacjenta i Rzeczniku Praw Pacjenta.
§ 20. [Przewożenie krwi i jej składników]
§ 21. [Przekazywanie krwi i jej składników]
1) zgodności etykiet z zamówieniem na krew i jej składniki;
2) daty ważności;
3) szczelności pojemników;
4) oceny wizualnej krwi lub jej składników.
2. Przyjęcie pojemników z krwią lub jej składnikami potwierdza się przez umieszczenie daty oraz czytelnego podpisu kierownika banku krwi lub osoby upoważnionej do odbioru na kopii kwitu rozchodowego centrum.
3. Przed wydaniem krwi lub jej składników do jednostek lub komórek organizacyjnych zakładu leczniczego należy dokładnie sprawdzić zgodność danych na etykiecie z zamówieniem na krew i jej składniki, w szczególności porównać numer składnika z numerem na wyniku próby zgodności, jeżeli obowiązuje jej wykonanie. W przypadku KKP, osocza oraz krioprecypitatu pracownik przed wydaniem tych składników krwi powinien sprawdzić grupę krwi pacjenta w dokumentacji pracowni immunologii transfuzjologicznej, a jeżeli w pracowni brak jest wyniku, należy zwrócić się do lekarza o przekazanie kopii wyniku z dokumentacji pacjenta. W sytuacjach nagłych, gdy lekarz zleca wydanie składników krwi do pilnego przetoczenia dla pacjenta, który nie ma oznaczonej grupy krwi, pracownik sprawdza, czy został wydany składnik o odpowiedniej grupie.
4. Krew i jej składniki powinny być wydawane do jednostek lub komórek organizacyjnych zakładu leczniczego bezpośrednio przed planowanym przetoczeniem po wykonaniu próby zgodności, jeżeli obowiązuje jej wykonanie, i innych wymaganych badań immunohematologicznych.
§ 22. [Zwrot krwi lub jej składników]
1) zgonu pacjenta, dla którego zamawiano krew lub jej składniki;
2) przypadku rzadko występującego fenotypu krwinek czerwonych;
3) innego uzasadnionego przypadku – po wyrażeniu zgody przez kierownika właściwego centrum;
4) uwzględnienia zgłoszonej reklamacji krwi lub jej składników.
2. Krew lub jej składniki można zwrócić w przypadku, o którym mowa w ust. 1, pod warunkiem że:
1) dana jednostka krwi lub jej składniki były przechowywane i transportowane we właściwy sposób przy zachowaniu odpowiedniej i prawidłowo kontrolowanej temperatury oraz przy użyciu sprzętu chłodniczego, w którym proces przechowywania został poddany walidacji;
2) w podmiocie leczniczym, z którego krew lub jej składniki są przyjmowane, właściwe centrum przeprowadziło wcześniej kontrolę potwierdzoną protokołem stwierdzającym brak uchybień w stosunku do obowiązujących przepisów dotyczących przechowywania krwi i jej składników.
3. Zwrot krwi lub jej składników może być przyjęty wyłącznie przez właściwe centrum na podstawie prawidłowo wypełnionego protokołu:
1) niewykorzystania krwi lub jej składników;
2) kontroli temperatury przechowywania krwi i jej składników;
3) kontroli temperatury transportu krwi i jej składników, który sporządza się w przypadku, gdy krew i jej składniki nie były przewożone środkami transportu kontrolowanymi przez centrum.
4. Protokoły, o których mowa w ust. 3, są sporządzane według wymagań określonych w obwieszczeniu wydanym na podstawie art. 24 pkt 2 lit. b ustawy.
§ 23. [Reklamacja w przypadku stwierdzenia nieprawidłowości przez odbierającego krew lub jej składniki]
2. W przypadku reklamacji należy przekazać do właściwego centrum bezpośrednio lub poprzez bank krwi reklamowaną krew lub jej składniki wraz z protokołem reklamacji, do którego załącza się:
1) protokół kontroli temperatury przechowywania krwi lub jej składników, jeżeli dotyczy;
2) protokół kontroli temperatury transportu krwi i jej składników (który sporządza się w przypadku, gdy krew i jej składniki nie były przewożone środkami transportu kontrolowanymi przez centrum).
3. Protokoły, o których mowa w ust. 2, muszą zawierać co najmniej dane opisane w obwieszczeniu wydanym na podstawie art. 24 pkt 2 lit. b ustawy.
4. Reklamacja może być rozpatrzona, pod warunkiem że:
1) reklamowana jednostka krwi lub jej składniki były przechowywane i transportowane we właściwy sposób przy zachowaniu odpowiedniej i prawidłowo kontrolowanej temperatury oraz przy użyciu sprzętu chłodniczego, w którym proces przechowywania został poddany walidacji;
2) w podmiocie leczniczym, od którego krew lub jej składniki są przyjmowane, właściwe centrum przeprowadziło wcześniej kontrolę, w wyniku której stwierdzono brak uchybień w stosunku do obowiązujących przepisów dotyczących przechowywania krwi i jej składników.
5. W przypadku uwzględnienia reklamacji dział ekspedycji właściwego centrum:
1) wydaje nieodpłatnie w miejsce zwróconych składników krwi inny, równoważny mu składnik krwi albo
2) nie nalicza opłaty za zwrócone składniki krwi.
Rozdział 4
Organizacja pracowni immunologii transfuzjologicznej w podmiocie leczniczym
§ 24. [Pracownia immunologii transfuzjologicznej]
1) odpowiednie pomieszczenie do wykonywania badań, w którym temperatura powinna wynosić 18–25°C;
2) odpowiedni sprzęt, aparaturę, odczynniki diagnostyczne, krwinki wzorcowe oraz odpowiednie formularze zleceń i wyników badań immunohematologicznych.
§ 25. [Kierownik pracowni immunologii transfuzjologicznej]
2. Kierownik pracowni immunologii transfuzjologicznej musi posiadać zaświadczenie upoważniające do samodzielnego wykonywania badań immunohematologicznych i autoryzowania ich wyników wydane przez kierownika jednostki organizacyjnej publicznej służby krwi, o której mowa w art. 4 ust. 3 ustawy, oraz co najmniej dwuletnią praktykę w wykonywaniu badań immunohematologicznych.
3. Kierownik organizuje pracę pracowni w sposób zapewniający gotowość do wykonywania badań immunohematologicznych przez całą dobę, gwarantując bezpieczeństwo biorców krwi, w tym noworodków zagrożonych chorobą hemolityczną wynikającą z konfliktu serologicznego.
4. Kierownik pracowni immunologii transfuzjologicznej sporządza SOP obowiązujące w pracowni, które zatwierdza kierownik podmiotu leczniczego.
§ 26. [Kwalifikacje do wykonywania badań immunohematologicznych]
2. Pracownik, o którym mowa w ust. 1, po co najmniej rocznej praktyce w wykonywaniu badań pod nadzorem osób wskazanych w ust. 1, odbywa dwutygodniowe szkolenie teoretyczne i praktyczne w jednostkach organizacyjnych publicznej służby krwi, o których mowa w art. 4 ust. 3 ustawy.
3. Po zdaniu egzaminu praktycznego i teoretycznego technik analityki medycznej otrzymuje zaświadczenie upoważniające do samodzielnego wykonywania badań immunohematologicznych, a lekarz lub diagnosta laboratoryjny otrzymuje zaświadczenie upoważniające do samodzielnego wykonywania badań immunohematologicznych i autoryzacji wyników.
4. Zaświadczenia są wydawane przez kierownika jednostki przeprowadzającej szkolenia. Wzór zaświadczenia upoważniającego do samodzielnego wykonywania badań immunohematologicznych oraz autoryzacji wyników dla lekarza i diagnosty laboratoryjnego oraz zaświadczenia upoważniającego do samodzielnego wykonywania badań immunohematologicznych dla technika analityki medycznej określa załącznik nr 12 do rozporządzenia.
5. Roczna praktyka w wykonywaniu badań pod nadzorem odbywa się w pełnym wymiarze czasu pracy lub niepełnym wymiarze czasu pracy odpowiadającym rocznemu pełnemu wymiarowi czasu pracy w okresie nie dłuższym niż trzy lata.
6. W przypadku przerwy w wykonywaniu pracy w pracowni immunologii transfuzjologicznej dłuższej niż 2 lata personel zatrudniony w pracowni immunologii transfuzjologicznej musi odbyć dodatkowe 14-godzinne szkolenie w jednostce organizacyjnej publicznej służby krwi, o której mowa w art. 4 ust. 3 ustawy.
7. Pracownia immunologii transfuzjologicznej co najmniej raz w roku jest poddawana kontroli jakości wykonywanych badań przeprowadzanej przez właściwe centrum.
8. Wynik dokonanej oceny jest przekazywany kierownikowi podmiotu leczniczego i kierownikowi pracowni immunologii transfuzjologicznej podmiotu leczniczego. W przypadku gdy przekazana ocena jest negatywna, kierownik podmiotu leczniczego podejmuje niezwłocznie działania mające na celu zapewnienie wymaganej jakości wykonywanych badań.
9. Liczba osób posiadających uprawnienia do samodzielnego wykonywania badań i autoryzacji wyników, zatrudnionych w pracowni immunologii transfuzjologicznej, jest uzależniona od zakresu i liczby wykonywanych badań.
10. Personel wykonujący badania immunohematologiczne w pracowni immunologii transfuzjologicznej nie może być kierowany na stanowiska pracy w innych pracowniach.
11. Wszystkie wyniki badań wydawane z pracowni immunologii transfuzjologicznej muszą być autoryzowane przez diagnostę laboratoryjnego lub lekarza posiadających zaświadczenie upoważniające do wykonywania badań i autoryzacji wyników w zakresie immunologii transfuzjologicznej.
12. Przebieg czynności wykonywanych w pracowni immunologii transfuzjologicznej poza godzinami z pełną obsadą pracowników pracowni, wynikającymi z organizacji czasu pracy pracowni immunologii transfuzjologicznej, zwanych dalej „pozaregulaminowymi godzinami pracy”, w tym w porze nocnej oraz w dniach ustawowo wolnych od pracy, należy odnotowywać w książce raportów. W raportach należy uwzględnić w szczególności napotkane trudności w interpretacji wyników badań, opis okoliczności związanej z wydaniem lub niewydaniem wyniku próby zgodności dla pacjenta, u którego wykryto nieregularne przeciwciała, oraz informację o zdalnej autoryzacji wyników badań, jeżeli była dokonana. Raporty podlegają codziennemu przeglądowi oraz analizie i są podpisywane przez kierownika pracowni immunologii transfuzjologicznej lub przez inną upoważnioną przez niego osobę.
13. Dopuszcza się zdalną autoryzację wyników badań wykonywaną zgodnie z warunkami, o których mowa w ust. 14–18.
14. Zdalnej autoryzacji może dokonać osoba, o której mowa w ust. 11, jeżeli jest zatrudniona lub wykonuje swoje zadania na innej podstawie niż stosunek pracy w pracowni, w której autoryzuje wyniki.
15. Osoba, o której mowa w ust. 11, może dokonywać zdalnej autoryzacji dla maksymalnie dwóch pracowni immunologii transfuzjologicznej znajdujących się na obszarze działania właściwego centrum, które wydało pozytywną opinię, o której mowa w ust. 18 pkt 6, dopuszczającą zdalną autoryzację.
16. Wyniki badań wydawane z pracowni immunologii transfuzjologicznej podmiotu leczniczego albo innego podmiotu wykonującego badania w zakresie krwiolecznictwa mogą być autoryzowane przez osoby, o których mowa w ust. 11, zatrudnione lub wykonujące swoje zadania na innej podstawie niż stosunek pracy w pracowni we właściwym centrum.
17. Zdalna autoryzacja wyników badań jest dopuszczalna wyłącznie w pozaregulaminowych godzinach pracy, w tym w porze nocnej oraz w dniach ustawowo wolnych od pracy.
18. Zdalnej autoryzacji wyników badań można dokonać, jeżeli:
1) badanie jest wykonywane za pomocą automatycznego analizatora immunohematologicznego;
2) osobie dokonującej zdalnej autoryzacji wyników badań zapewniono:
a) dostęp do danych operacyjnych, protokołów badań zapisanych w programie komputerowym oraz do obrazów reakcji pobranych z analizatora z możliwością wprowadzenia zmian w razie takiej konieczności, historii badań pacjentów oraz wyników kontroli jakości badań przeprowadzanych codziennie,
b) interaktywną komunikację audiowizualną umożliwiającą monitorowanie przebiegu badania od przyjęcia próbki do wydania wyniku;
3) pracownia immunologii transfuzjologicznej posiada oprogramowanie analizatora, o którym mowa w pkt 1, które jest zintegrowane z programem pracowni immunologii transfuzjologicznej;
4) pracownia immunologii transfuzjologicznej posiada szyfrowany bezpieczny dostęp do bazy danych zapewniony przez bezpieczne łącze internetowe;
5) osoba dokonująca zdalnej autoryzacji wyniku badania stosuje kwalifikowany podpis elektroniczny, zaawansowany podpis elektroniczny w rozumieniu art. 3 pkt 11 rozporządzenia Parlamentu Europejskiego i Rady (UE) nr 910/2014 z dnia 23 lipca 2014 r. w sprawie identyfikacji elektronicznej i usług zaufania w odniesieniu do transakcji elektronicznych na rynku wewnętrznym oraz uchylającego dyrektywę 1999/93/WE (Dz. Urz. UE L 257 z 28.08.2014, s. 73) albo podpis potwierdzony profilem zaufanym ePUAP w rozumieniu art. 3 pkt 15 ustawy z dnia 17 lutego 2005 r. o informatyzacji działalności podmiotów realizujących zadania publiczne (Dz. U. z 2017 r. poz. 570);
6) w pracowni immunologii transfuzjologicznej przeprowadzono walidację procesów wpływających na prawidłowy przebieg autoryzacji wyników i uzyskano pozytywną opinię właściwego centrum oraz instytutu, o którym mowa w art. 4 ust. 3 pkt 1 ustawy, w tym zakresie.
§ 27. [Wyposażenie pracowni immunologii transfuzjologicznej]
§ 28. [Nadzór specjalistyczny nad pracowniami immunologii transfuzjologicznej]
2. W ramach nadzoru właściwe centrum:
1) opiniuje liczbę niezbędnych do zatrudnienia osób w zależności od zakresu i liczby wykonywanych badań;
2) szkoli pracowników pracowni przed dopuszczeniem do samodzielnego wykonywania badań i autoryzacji wyników;
3) wydaje zaświadczenia uprawniające do samodzielnego wykonywania badań i autoryzacji wyników;
4) przeprowadza dodatkowe szkolenia dla pracowników w przypadku przerwy w wykonywaniu badań dłuższej niż 2 lata;
5) przeprowadza, co najmniej raz na dwa lata, okresowe kontrole organizacji pracy, stosowanych metod i procedur oraz wyposażenia i warunków pracy;
6) wydaje zalecenia pokontrolne oraz nadzoruje ich wykonanie w przypadku stwierdzenia nieprawidłowości;
7) zaleca wprowadzenie uzasadnionych zmian w funkcjonowaniu pracowni oraz modyfikację metod badań immunohematologicznych i dokumentacji;
8) zaleca przeprowadzenie dodatkowego szkolenia pracowników, w przypadku gdy przyczyną niepożądanego zdarzenia lub niepożądanej reakcji był błąd pracownika pracowni immunologii transfuzjologicznej;
9) w przypadku niezrealizowania przez kontrolowany podmiot, w wyznaczonym terminie, zaleceń, o których mowa w pkt 6, powiadamia wojewodę właściwego dla siedziby kontrolowanego podmiotu leczniczego.
3. Protokół z kontroli przeprowadzonej przez właściwe centrum przekazuje się kierownikowi podmiotu leczniczego i kierownikowi pracowni immunologii transfuzjologicznej.
4. Właściwe centrum przekazuje do instytutu, o którym mowa w art. 4 ust. 3 pkt 1 ustawy, corocznie, do dnia 31 marca, podsumowanie wyników kontroli przeprowadzonych w pracowniach immunologii transfuzjologicznej.
§ 29. [Przyjmowanie próbek krwi do badań]
2. W przypadku braku danych pacjenta do badania, można przyjąć próbki krwi i zlecenia z wpisanym symbolem „NN”, płcią lub niepowtarzalnym numerem identyfikacyjnym pacjenta oraz resortowym kodem identyfikacyjnym podmiotu zlecającego badanie, o którym mowa w przepisach wydanych na podstawie art. 105 ust. 5 ustawy z dnia 15 kwietnia 2011 r. o działalności leczniczej.
3. Po otrzymaniu próbek krwi należy sprawdzić zgodność danych na etykietach probówek i zleceniach. Jeżeli dane na probówce są niezgodne z danymi na zleceniu, należy wyjaśnić przyczynę rozbieżności i w razie potrzeby zażądać ponownego pobrania próbki krwi od pacjenta. Niezgodności takie należy monitorować i dokumentować w sposób przewidziany dla niepożądanych zdarzeń lub reakcji.
4. Próbki krwi należy zarejestrować w książce badań grup krwi, której wzór jest określony w załączniku nr 13 do rozporządzenia, lub w książce prób zgodności, której wzór jest określony w załączniku nr 14 do rozporządzenia, albo w systemie informatycznym, nadając im kolejne numery przy zachowaniu ciągłości numeracji w danym roku.
5. W przypadku biorców krwi leczonych krwią w przeszłości oraz kobiet, u których w wyniku przeprowadzonego wywiadu medycznego uzyskano informację o przebytych ciążach, należy zapoznać się z całą dostępną dokumentacją dotyczącą poszukiwania i identyfikacji przeciwciał, a także dokumentacją dotyczącą niepożądanych reakcji, jeżeli takie wystąpiły.
6. Próbki krwi pacjentów i dawców krwi przechowuje się w temperaturze od 2°C do 8°C przez 72 godziny.
§ 30. [Zakres badań wykonywanych w pracowni immunologii transfuzjologicznej]
1) określanie grup krwi ABO i RhD oraz przeglądowe badanie na obecność przeciwciał odpornościowych do antygenów krwinek czerwonych;
2) wykonywanie prób zgodności serologicznej krwi.
2. Do rozszerzonego zakresu badań wykonywanych w pracowni immunologii transfuzjologicznej należy:
1) badanie w kierunku konfliktu serologicznego między matką a płodem;
2) badanie kwalifikujące do podania immunoglobuliny anty-RhD w ramach profilaktyki konfliktu serologicznego RhD;
3) badanie w celu identyfikacji przeciwciał;
4) określanie antygenu K oraz innych antygenów grupowych krwi.
3. W przypadku konieczności pilnego przetoczenia i trudności w oznaczeniu grupy krwi ABO lub RhD oraz w przypadku braku wiarygodnego wyniku grupy krwi, o którym mowa w § 9 ust. 2, jeżeli lekarz zdecyduje o przetoczeniu, dopuszcza się do czasu otrzymania wyniku grupy krwi wydanie składników krwi:
1) KKCz grupy O przed wykonaniem próby zgodności, a w przypadku pacjentów z alloprzeciwciałami anty-D oraz dziewczynek i kobiet do okresu menopauzy O RhD ujemny K ujemny;
2) osocza lub krioprecypitatu grupy AB;
3) KKP – składnik grupy O zawieszony w osoczu grupy AB lub w odpowiednim roztworze wzbogacającym lub KKP grupy AB.
4. Przy braku KKCz O RhD ujemnego dopuszcza się przetoczenie dziewczynkom i kobietom do okresu menopauzy KKCz grupy O RhD dodatniego.
5. W celu optymalnego wykorzystania krwi grupy O należy jak najszybciej po otrzymaniu wyniku grupy krwi przetaczać KKCz zgodne z oznaczoną grupą.
6. W przypadku pacjentów, u których uzyskano dodatni wynik badania przeglądowego, dokonuje się identyfikacji przeciwciał. W przypadku konieczności pilnego przetoczenia dopuszcza się przetoczenie KKCz przed otrzymaniem wyniku identyfikacji przeciwciał, opierając się na zgodności krwi w ABO i RhD oraz ujemnym wyniku reakcji surowicy pacjenta z krwinkami dawcy, czyli na ujemnym wyniku w próbie krzyżowej. Wynik należy sformułować: „Krew dawcy nr .... zgodna w próbie krzyżowej”. Krew można przetoczyć w sytuacji bezpośredniego zagrożenia życia pacjenta. W uwagach należy dopisać: „W surowicy pacjenta wykryto alloprzeciwciała. Identyfikacja w toku”.
7. Jeżeli pracownia immunologii transfuzjologicznej nie ma możliwości wykonania identyfikacji przeciwciał, przesyła próbki do jednostki organizacyjnej publicznej służby krwi wraz ze zleceniem na konsultacyjne badania immunohematologiczne, którego wzór jest określony w załączniku nr 15 do rozporządzenia, i protokołem wykonanych badań.
8. Właściwe centrum wydaje wynik badania w trzech egzemplarzach, z których jeden egzemplarz jest przeznaczony dla pracowni immunologii transfuzjologicznej podmiotu leczniczego w celu uzupełnienia dokumentacji wyników badań, jeden egzemplarz dla pacjenta i jeden egzemplarz dla lekarza w celu umieszczenia w historii choroby.
9. W przypadkach pilnego przetoczenia dopuszcza się przetoczenie KKCz przed otrzymaniem wyniku identyfikacji przeciwciał, opierając się na zgodności krwi w ABO i RhD oraz ujemnym wyniku reakcji surowicy pacjenta z krwinkami dawcy – ujemnym wyniku w próbie krzyżowej.
10. U biorców krwi systematycznie leczonych KKCz oraz u osób, którym przetaczano KKCz w okresie ostatnich 3 miesięcy, bezwzględnie należy przestrzegać czasu ważności próby zgodności, który – liczony od momentu pobrania próbki krwi od pacjenta – wynosi 48 godzin. Jeżeli KKCz nie został w tym czasie przetoczony, należy powtórnie wykonać próbę zgodności ze świeżo pobranej próbki krwi od pacjenta.
11. Próbę zgodności wykonuje się z próbki krwi biorcy krwi pobranej wyłącznie w tym celu. Jako krew dawcy służy próbka zawarta w segmencie drenu połączonego z pojemnikiem z KKCz, KPK lub KG. Przed odłączeniem segmentu drenu należy porównać jego numer donacji z numerem na etykiecie pojemnika.
12. Wyniki próby zgodności wpisuje się na formularzu wyniku próby zgodności.
13. Wzór zlecenia na wykonanie badań immunohematologicznych kwalifikujących do podania immunoglobuliny anty-RhD w ramach profilaktyki konfliktu serologicznego RhD jest określony w załączniku nr 16 do rozporządzenia. Wzór wyniku tych badań jest określony w załączniku nr 17 do rozporządzenia.
§ 31. [Dokumentacja i autoryzacja badań wykonywanych w pracowni immunologii transfuzjologicznej]
2. Badanie jest zakończone sporządzeniem wyniku, który podpisuje osoba wykonująca to badanie i osoba autoryzująca wynik.
3. W pozaregulaminowych godzinach pracy, w tym w porze nocnej oraz w dniach ustawowo wolnych od pracy, może być wydany wynik badania wykonany przez osobę uprawnioną do wykonywania i autoryzowania wyników i jednocześnie przez nią podpisany jako osobę wykonującą badanie i autoryzującą wynik.
4. Dopuszcza się podpisanie wyniku w sposób określony w przepisach wydanych na podstawie art. 30 ust. 1 ustawy z dnia 6 listopada 2008 r. o prawach pacjenta i Rzeczniku Praw Pacjenta i przekazanie go drogą elektroniczną do oddziału lub kliniki.
5. Jeżeli badania wykonuje się metodą automatyczną, dopuszcza się prowadzenie dokumentacji w postaci wydruków komputerowych zamiast książek, o których mowa w ust. 1, przy czym wydruki muszą uwzględniać wszystkie dane ujęte we wzorach tych książek.
§ 32. [Dobór krwi i jej składników]
2. Pacjentom, u których wykryto autoprzeciwciała do krwinek czerwonych reagujące w temperaturze 37°C, należy dobierać krew lub jej składniki zgodne fenotypowo w układzie Rh i antygenie K z układu Kell.
3. Dla kobiet do okresu menopauzy jest wskazane dobieranie KKCz zgodnego w układzie ABO i RhD oraz dodatkowo K ujemnego w ramach profilaktyki konfliktu serologicznego w antygenie K. Jeżeli wykonano badania antygenu K u pacjenta i stwierdzono jego obecność, można przetaczać KKCz K dodatni.
4. Biorcom krwi, u których wykryto przeciwciała odpornościowe w aktualnym badaniu lub w przeszłości, dobiera się KKCz niezawierający odpowiadającego im antygenu oraz zgodny fenotypowo w układzie Rh i antygenie K z układu Kell.
5. Dopuszcza się przetaczanie KKCz grupy O pacjentom innej grupy krwi, w przypadku:
1) braku krwi jednoimiennej w układzie ABO;
2) bardzo słabej ekspresji antygenu A lub B albo trudności w oznaczeniu grupy krwi ABO.
6. Dopuszcza się przetaczanie KKCz grupy A lub B biorcom grupy AB, w przypadku braku krwi jednoimiennej.
7. Biorcy RhD dodatniemu można przetaczać KPK, KKCz, KG i KKP RhD dodatni lub RhD ujemny.
8. W przypadku bezpośredniego zagrożenia życia, na pisemne zamówienie na krew lub jej składniki do pilnego przetoczenia wydane przez lekarza prowadzącego leczenie lub lekarza odpowiedzialnego za gospodarkę krwią, można wydać krew i jej składniki zgodne w układzie ABO i RhD z biorcą przed wykonaniem próby zgodności. Wydanie KKCz zgodnego w ABO i RhD z biorcą jest możliwe wyłącznie na podstawie dwóch jednobrzmiących wyników grupy krwi ABO i RhD, wpisu w karcie identyfikacyjnej grupy krwi albo w legitymacji żołnierza.
9. Po wydaniu KKCz do pilnego przetoczenia zgodnie z ust. 8 przystępuje się niezwłocznie do wykonania próby zgodności. Jeżeli wynik wskazuje na niezgodność, jest konieczne natychmiastowe powiadomienie o tym lekarza prowadzącego leczenie w celu przerwania przetoczenia.
10. W wyjątkowo nagłym przypadku pilnego przetoczenia, jeżeli lekarz prowadzący leczenie zdecyduje o przetoczeniu przed wykonaniem badania grupy krwi u biorcy krwi i próby zgodności, należy wydać:
1) KKCz grupy O, a kobietom w wieku rozrodczym – KKCz grupy O RhD ujemny, K ujemny;
2) osocze grupy AB;
3) krioprecypitat grupy AB;
4) KKP grupy O zawieszone w osoczu grupy AB lub w roztworze wzbogacającym; dopuszcza się także przetoczenie KKP grupy AB, jeżeli rekonstytuowany KKP jest niedostępny.
11. W przypadku, o którym mowa w ust. 10, należy natychmiast przystąpić do określenia u biorcy grupy krwi ABO i RhD, wykonania przeglądowego badania przeciwciał odpornościowych i próby zgodności. Do dalszych przetoczeń należy kwalifikować krew lub jej składniki jednoimienne z biorcą w układzie ABO i RhD.
§ 33. [Przetaczanie u noworodków i niemowląt]
1) określić grupę krwi w układzie ABO i antygen D z układu Rh u matki i u dziecka;
2) wykonać badania w kierunku obecności alloprzeciwciał odpornościowych w surowicy matki;
3) wykonać bezpośredni test antyglobulinowy, zwany dalej „BTA”, u dziecka.
2. Jeżeli w surowicy matki nie wykrywa się alloprzeciwciał odpornościowych i BTA dziecka jest ujemny:
1) noworodkom i niemowlętom do ukończenia 4 miesiąca życia urodzonym przez matki tej samej grupy krwi ABO co dziecko przetacza się KKCz zgodne w ABO i RhD dziecka bez wykonywania próby krzyżowej krwinek dawcy z surowicą matki, po sprawdzeniu zgodności oznaczenia antygenów A, B i D dawców przeznaczonych do przetoczenia; można również przetoczyć KKCz grupy O;
2) noworodkom i niemowlętom do ukończenia 4 miesiąca życia urodzonym przez matki innej grupy krwi ABO niż dziecko przetacza się KKCz grupy O i RhD zgodnym z RhD dziecka bez wykonywania próby krzyżowej krwinek dawcy z surowicą matki, po sprawdzeniu zgodności oznaczenia antygenów A, B i D dawców przeznaczonych do przetoczenia;
3) w przypadku gdy matka jest grupy AB, dziecku można przetoczyć KKCz grupy krwi dziecka lub KKCz grupy O, RhD zgodne z RhD dziecka bez wykonywania próby krzyżowej krwinek dawcy z surowicą matki, po sprawdzeniu zgodności oznaczenia antygenów A, B i D dawców przeznaczonych do przetoczenia.
3. Jeżeli w surowicy matki wykrywa się alloprzeciwciała odpornościowe spoza układu ABO:
1) noworodkom i niemowlętom do ukończenia 4 miesiąca życia urodzonym przez matki tej samej grupy krwi ABO dobiera się KKCz zgodne w grupie ABO oraz bez antygenu, do którego są skierowane alloprzeciwciała; przed każdym przetoczeniem należy wykonywać próbę krzyżową z surowicą matki;
2) noworodkom i niemowlętom do ukończenia 4 miesiąca urodzonym przez matki innej grupy krwi ABO dobiera się KKCz grupy O bez antygenu, do którego są skierowane alloprzeciwciała; przed każdym przetoczeniem należy wykonywać próbę krzyżową z surowicą matki.
4. Jeżeli krew matki jest niedostępna, należy sprawdzić obecność odpornościowych przeciwciał w surowicy dziecka oraz BTA na krwinkach dziecka, a w przypadku gdy:
1) nie wykrywa się przeciwciał odpornościowych i BTA jest ujemny – należy przetaczać KKCz grupy O, RhD zgodne z dzieckiem i wykonać próbę krzyżową z surowicą dziecka;
2) BTA jest dodatni i w surowicy dziecka wykryto alloprzeciwciała odpornościowe – należy ustalić ich swoistość i do przetoczenia należy dobierać krwinki grupy O ujemne w antygenie, do którego jest skierowana swoistość przeciwciał; należy zawsze wykonać próbę krzyżową z surowicą dziecka; można użyć eluatu z jego krwinek.
5. Ujemny wynik badania przeglądowego na obecność przeciwciał i ujemny wynik w próbie krzyżowej upoważniają do odstąpienia od wykonywania tych badań przed następnymi przetoczeniami.
6. Dodatni wynik badania przeglądowego na obecność przeciwciał nakazuje wykonywanie próby krzyżowej z surowicą lub osoczem dziecka.
§ 34. [Dobieranie KKCz do przetaczania dopłodowego]
1) należy dobierać krwinki czerwone grupy O niezawierające antygenów, do których są skierowane przeciwciała wykryte u matki;
2) w celu uniknięcia uodpornienia ciężarnej antygenami zawartymi w KKCz użytym do przetoczenia dopłodowego należy oznaczyć u niej antygeny z układów grupowych Rh (D, C, Cw, c, E, e), Kell (K i k), Kidd (Jka i Jkb), Duffy (Fya i Fyb) i MNS (S i s) oraz dobierać do przetoczenia krwinki czerwone dawców zgodne z krwią matki;
3) jeżeli grupa krwi ABO płodu jest zgodna z grupą krwi matki oraz gdy matka ma grupę AB, a płód ma grupę A lub B, można dobierać krwinki jednoimienne w układzie ABO z dzieckiem.
§ 35. [Dobieranie krwi do przetoczenia wymiennego]
1) w konflikcie RhD należy dobierać krwinki czerwone RhD ujemne, przy czym:
a) w przypadku gdy zachodzi zgodność serologiczna w układzie ABO między matką i dzieckiem, przetacza się krew pełną rekonstytuowaną, zwaną dalej „KPR”, składającą się z KKCz i osocza od dawców o zgodnej grupie krwi w układzie ABO z dzieckiem,
b) noworodkom o grupie krwi A lub B matek innej grupy krwi w układzie ABO niż dziecko przetacza się KPR, składającą się z KKCz od dawcy grupy O, zawieszonego w osoczu od dawcy grupy zgodnej z grupą krwi dziecka lub w osoczu grupy AB,
c) noworodkom urodzonym przez matkę grupy AB przetacza się KPR składający się z KKCz i osocza grupy krwi ABO zgodnej z dzieckiem;
2) w konflikcie w układzie ABO przetacza się KPR składającą się z KKCz od dawcy grupy O i RhD zgodnym z RhD dziecka, zawieszonego w osoczu od dawcy grupy krwi ABO zgodnej z grupą krwi dziecka lub w osoczu grupy AB;
3) w konflikcie serologicznym w zakresie innych antygenów niż wymienione w pkt 1 i 2 KKCz przeznaczony do sporządzenia KPR nie może zawierać antygenu odpowiedzialnego za uodpornienie matki.
2. W okresie pierwszych 4 miesięcy życia dziecka do kolejnych przetoczeń uzupełniających stosuje się KKCz tej samej grupy ABO i RhD, jak do pierwszego przetoczenia.
§ 36. [Trwałe i wiarygodne udokumentowania wyniku grupy krwi]
§ 37. [Stosowanie przepisów rozporządzenia]
Rozdział 5
Monitorowanie i zgłaszanie niepożądanych zdarzeń i niepożądanych reakcji
§ 38. [Procedura w przypadku poważnych niepożądanych reakcji i poważnych niepożądanych zdarzeń]
2. Poważne niepożądane reakcje, o których mowa w § 14 ust. 1 i 2, oraz poważne niepożądane zdarzenia zgłasza się na formularzu zgłoszenia niepożądanej reakcji lub zdarzenia, o którym mowa w załączniku nr 10 do rozporządzenia.
3. Do właściwego centrum przesyła się raporty dotyczące niepożądanych reakcji i niepożądanych zdarzeń. Właściwe centrum dla podmiotu leczniczego rejestruje wszystkie niepożądane reakcje i zdarzenia na podstawie otrzymanych raportów i uwzględnia je w danych statystycznych.
4. Podmiot leczniczy rejestruje także niepożądane zdarzenia, które mogły zagrażać bezpieczeństwu pacjenta, ale były usunięte przed przetoczeniem, i raporty o nich przesyła do właściwego centrum.
5. Jeżeli aktualne badanie dawcy wykazało potwierdzoną obecność zakażenia wirusami HBV, HCV lub HIV, właściwe centrum rozpoczyna procedurę prześledzenia drogi krwi od dawcy do biorcy w celu ustalenia biorców krwi, którzy mogli ulec zakażeniu tymi wirusami w okresie wczesnego zakażenia dawcy, w którym pomimo obecności czynników zakaźnych jeszcze się ich nie wykrywa stosowanymi metodami (okienko diagnostyczne).
6. Lekarz, który sprawował opiekę nad pacjentem, u którego wystąpiło podejrzenie poprzetoczeniowego zakażenia jednym z wirusów HBV, HCV lub HIV, lub lekarz wyznaczony przez kierownika jednostki lub komórki organizacyjnej podmiotu leczniczego ma obowiązek poinformować o tym pacjenta i zlecić odpowiednie badania w celu potwierdzenia lub wykluczenia zakażenia. Podmiot leczniczy informuje właściwe centrum o rezultatach przeprowadzonej procedury – również w przypadku, gdy badania nie zostały wykonane – wraz z podaniem przyczyny niewykonania badań.
7. Jeżeli u biorcy rozpoznano TRALI, właściwe centrum rozpoczyna procedurę prześledzenia drogi krwi od biorcy do dawcy w celu stwierdzenia, czy krew i jej składniki od tego samego dawcy spowodowały wymienione niepożądane reakcje u innych biorców krwi.
Rozdział 6
Przepisy przejściowe i przepis końcowy
§ 39. [Kierownicy pracowni immunologii transfuzjologicznej]
1) osoba zatrudniona na tym stanowisku w dniu wejścia w życie rozporządzenia nieposiadająca kwalifikacji, o których mowa w § 25 ust. 1 i 2,
2) diagnosta laboratoryjny lub lekarz niezatrudniony w dniu wejścia w życie rozporządzenia na stanowisku kierownika, jeżeli posiada co najmniej 3-letnie doświadczenie zawodowe w wykonywaniu badań immunohematologicznych
– pod warunkiem, że osoba ta posiada zaświadczenie uprawniające do wykonywania badań i autoryzacji wyników i jest w trakcie specjalizacji w dziedzinie laboratoryjnej transfuzjologii medycznej lub transfuzjologii klinicznej.
§ 40. [Powierzanie obowiązków lekarza odpowiedzialnego za gospodarkę krwią i jej składnikami]
§ 41. [Komitety transfuzjologiczne i lekarze odpowiedzialni za gospodarkę krwią powołani na podstawie dotychczasowych przepisów]
2. Lekarze odpowiedzialni za gospodarkę krwią powołani na podstawie dotychczasowych przepisów z dniem wejścia w życie niniejszego rozporządzenia stają się lekarzami odpowiedzialnym za gospodarkę krwią w rozumieniu niniejszego rozporządzenia.
§ 42. [Zaświadczenia upoważniające do wykonywania badań serologicznych uzyskane przed wejściem w życie rozporządzenia]
§ 43. [Wejście w życie]
Minister Zdrowia: K. Radziwiłł
1) Minister Zdrowia kieruje działem administracji rządowej - zdrowie, na podstawie § 1 ust. 2 rozporządzenia Prezesa Rady Ministrów z dnia 17 listopada 2015 r. w sprawie szczegółowego zakresu działania Ministra Zdrowia (Dz. U. poz. 1908).
2) Niniejsze rozporządzenie w zakresie swojej regulacji wdraża:
1) dyrektywę 2002/98/WE Parlamentu Europejskiego i Rady z dnia 27 stycznia 2003 r. ustanawiającą normy jakości i bezpiecznego pobierania, badania, preparatyki, przechowywania, wydawania krwi ludzkiej i składników krwi oraz zmieniającej dyrektywę 2001/83/WE (Dz. Urz. UE L 33 z 08.02.2003, str. 30 - Dz. Urz. UE Polskie wydanie specjalne, rozdz. 15, t. 7, str. 346, Dz. Urz. UE L 230 z 24.08.2006, str. 12, Dz. Urz. UE L 188 z 18.07.2009, str. 14 oraz Dz. Urz. UE L 98 z 15.04.2015, str. 11);
2) dyrektywę Komisji 2004/33/WE z dnia 22 marca 2004 r. wykonującą dyrektywę 2002/98/WE Parlamentu Europejskiego i Rady w zakresie niektórych wymagań technicznych dotyczących krwi i składników krwi (Dz. Urz. UE L 91 z 30.03.2004, str. 25 - Dz. Urz. UE Polskie wydanie specjalne, rozdz. 15, t. 8, str. 272, Dz. Urz. UE L 288 z 04.11.2009, str. 7, Dz. Urz. UE L 97 z 12.04.2011, str. 28 oraz Dz. Urz. UE L 366 z 20.12.2014, str. 81);
3) dyrektywę Komisji 2005/61/WE z dnia 30 września 2005 r. wykonującą dyrektywę 2002/98/WE Parlamentu Europejskiego i Rady w zakresie wymogów dotyczących śledzenia losów krwi oraz powiadamiania o poważnych, niepożądanych reakcjach i zdarzeniach (Dz. Urz. UE L 256 z 01.10.2005, str. 32);
4) dyrektywę Komisji 2005/62/WE z dnia 30 września 2005 r. wykonującą dyrektywę 2002/98/WE Parlamentu Europejskiego i Rady w zakresie norm i specyfikacji wspólnotowych odnoszących się do systemu jakości obowiązującego w placówkach służby krwi (Dz. Urz. UE L 256 z 01.10.2005, str. 41 oraz Dz. Urz. UE L 199 z 26.07.2016, str. 14).
3) Niniejsze rozporządzenie było poprzedzone rozporządzeniem Ministra Zdrowia z dnia 11 grudnia 2012 r. w sprawie leczenia krwią w podmiotach leczniczych wykonujących działalność leczniczą w rodzaju stacjonarne i całodobowe świadczenia zdrowotne, w których przebywają pacjenci ze wskazaniami do leczenia krwią i jej składnikami (Dz. U. z 2013 r. poz. 5), które utraciło moc z dniem 12 września 2017 r. zgodnie z art. 15 ust. 1 ustawy z dnia 20 maja 2016 r. o zmianie ustawy o publicznej służbie krwi oraz niektórych innych ustaw (Dz. U. poz. 823 oraz z 2017 r. poz. 1524).
Załączniki do rozporządzenia Ministra Zdrowia
z dnia 16 października 2017 r. (poz. 2051)
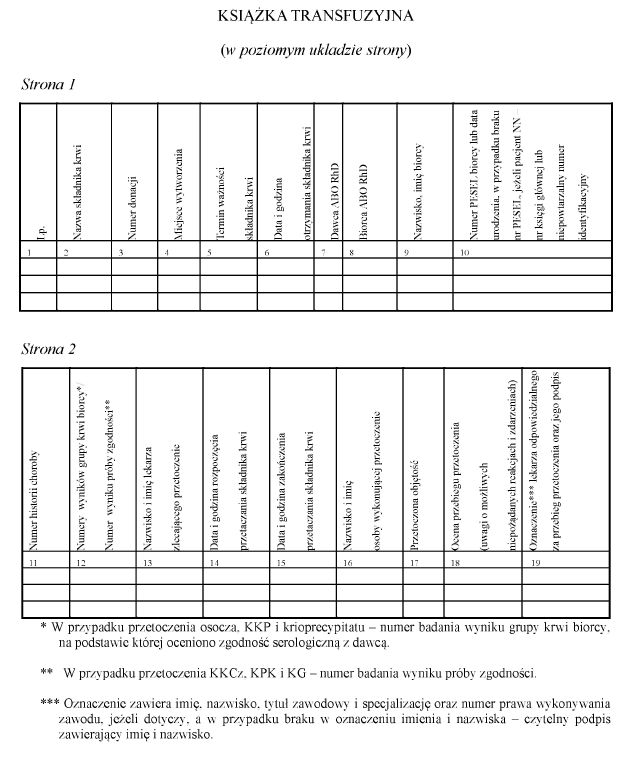
Załącznik nr 1
WZÓR – KSIĄŻKA TRANSFUZYJNA
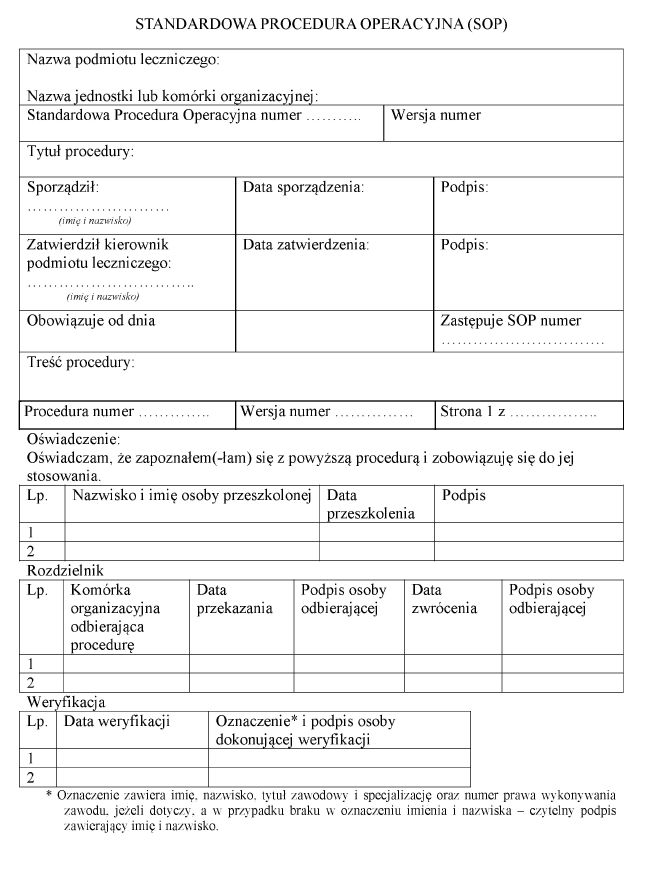
Załącznik nr 2
WZÓR – STANDARDOWA PROCEDURA OPERACYJNA (SOP)
Załącznik nr 3
WZÓR – ZAMÓWIENIE INDYWIDUALNE NA KREW LUB JEJ SKŁADNIKI

Załącznik nr 4
WZÓR – KARTA IDENTYFIKACYJNA GRUPY KRWI

Załącznik nr 5
WZÓR – ZLECENIE NA BADANIE GRUPY KRWI

Załącznik nr 6
WZÓR – ZLECENIE NA WYKONANIE PRÓBY ZGODNOŚCI

Załącznik nr 7
WZÓR – WYNIK BADANIA GRUPY KRWI


Załącznik nr 8
WZÓR – ZAMÓWIENIE NA KREW I JEJ SKŁADNIKI DO PILNEGO PRZETOCZENIA


Załącznik nr 9
WZÓR – WYNIK PRÓBY ZGODNOŚCI


Załącznik nr 10
WZÓR – ZGŁOSZENIE NIEPOŻĄDANEJ REAKCJI LUB ZDARZENIA

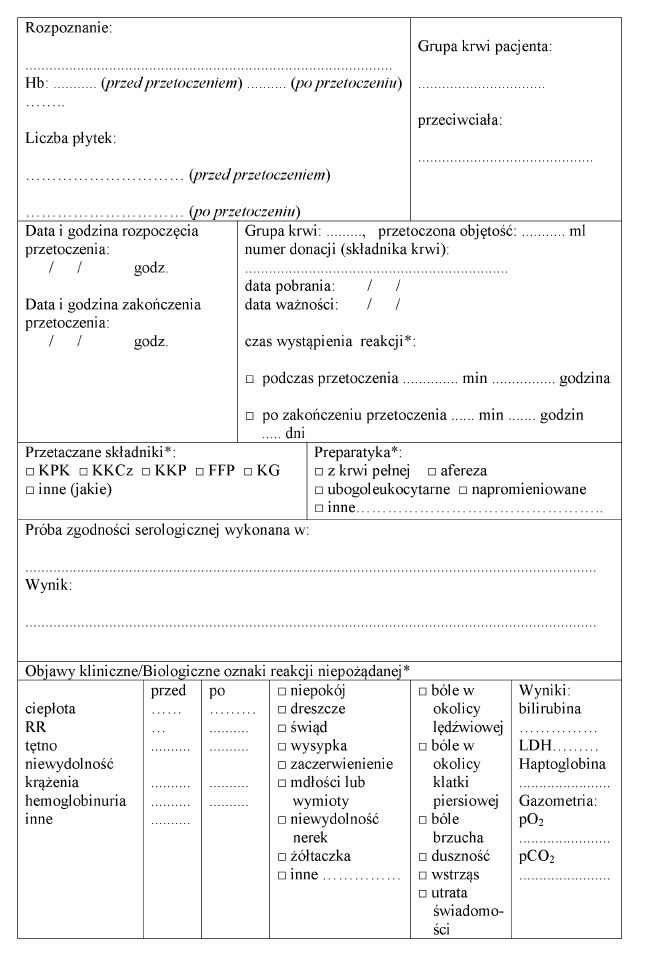


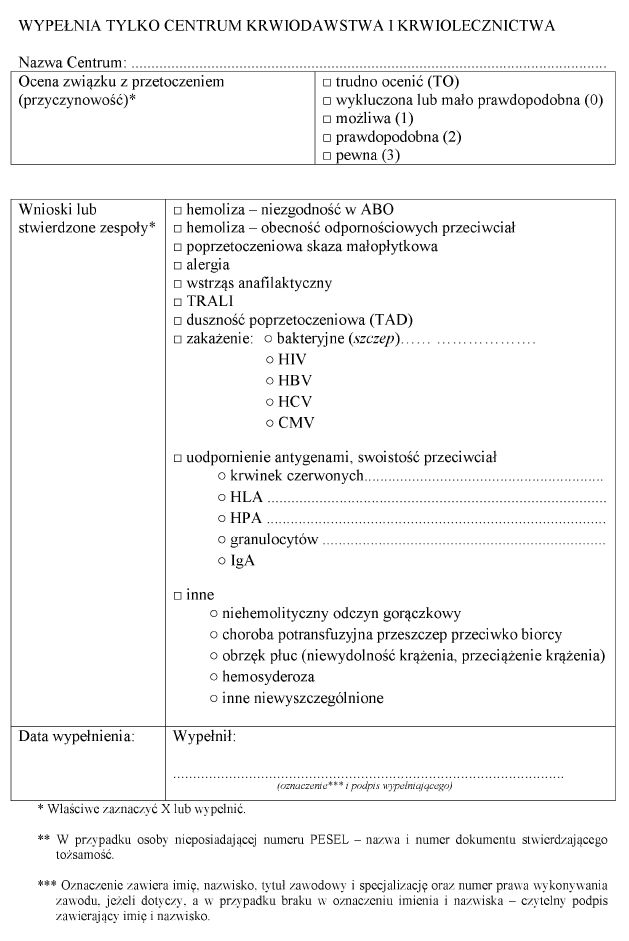
Załącznik nr 11
WZÓR – ZAMÓWIENIE ZBIORCZE NA KREW LUB JEJ SKŁADNIKI

Załącznik nr 12
WZÓR – ZAŚWIADCZENIE UPOWAŻNIAJĄCE DO SAMODZIELNEGO WYKONYWANIA BADAŃ IMMUNOHEMATOLOGICZNYCH ORAZ AUTORYZACJI WYNIKÓW DLA DIAGNOSTY LABORATORYJNEGO I LEKARZA ORAZ ZAŚWIADCZENIE UPOWAŻNIAJĄCE DO SAMODZIELNEGO WYKONYWANIA BADAŃ IMMUNOHEMATOLOGICZNYCH DLA TECHNIKA ANALITYKI MEDYCZNEJ


Załącznik nr 13
WZÓR – KSIĄŻKA BADAŃ GRUP KRWI
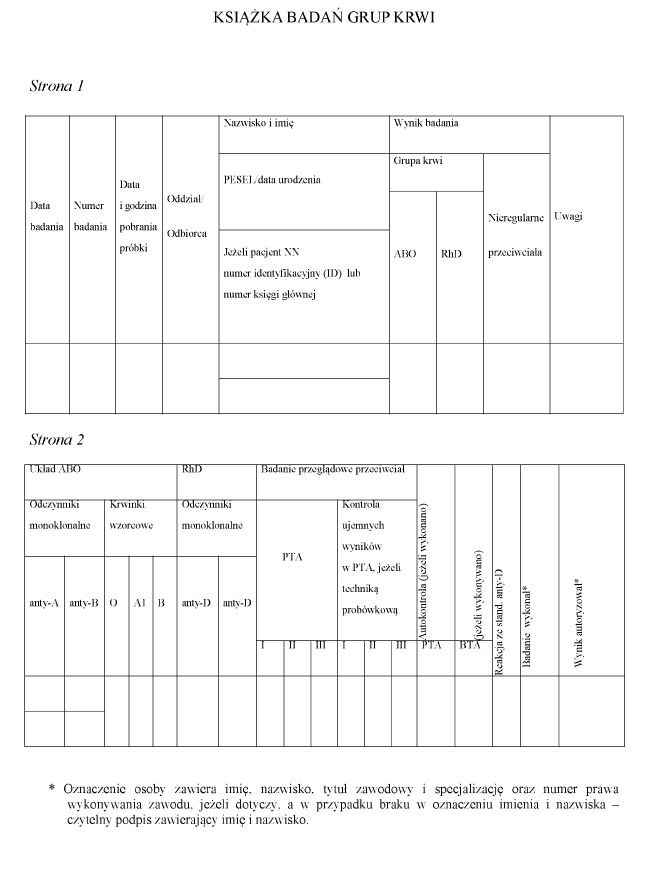
Załącznik nr 14
WZÓR – KSIĄŻKA PRÓB ZGODNOŚCI
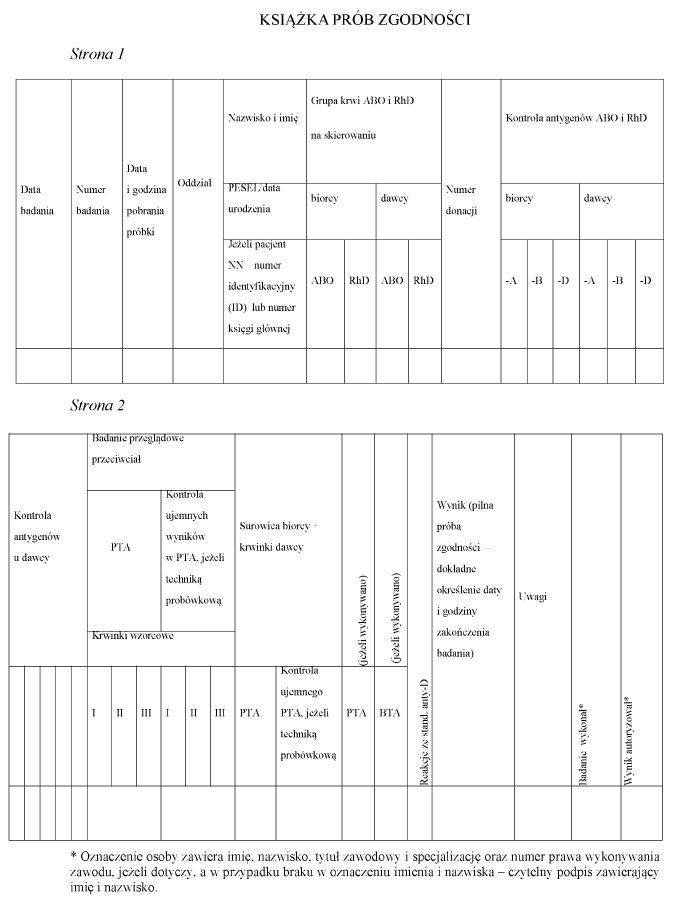
Załącznik nr 15
WZÓR – ZLECENIE NA KONSULTACYJNE BADANIE IMMUNOHEMATOLOGICZNE


Załącznik nr 16
WZÓR – ZLECENIE NA WYKONANIE BADAŃ IMMUNOHEMATOLOGICZNYCH KWALIFIKUJĄCYCH DO PODANIA IMMUNOGLOBULINY ANTY-D W RAMACH PROFILAKTYKI KONFLIKTU SEROLOGICZNEGO RhD


Załącznik nr 17
WZÓR – WYNIK BADAŃ IMMUNOHEMATOLOGICZNYCH KWALIFIKUJĄCYCH DO PODANIA IMMUNOGLOBULINY ANTY-D


- Data ogłoszenia: 2017-11-06
- Data wejścia w życie: 2017-11-07
- Data obowiązywania: 2020-03-27
- Dokument traci ważność: 2021-03-18

- ROZPORZĄDZENIE MINISTRA ZDROWIA z dnia 8 lipca 2019 r. zmieniające rozporządzenie w sprawie leczenia krwią i jej składnikami w podmiotach leczniczych wykonujących działalność leczniczą w rodzaju stacjonarne i całodobowe świadczenia zdrowotne
- ROZPORZĄDZENIE MINISTRA ZDROWIA z dnia 26 marca 2020 r. zmieniające rozporządzenie w sprawie leczenia krwią i jej składnikami w podmiotach leczniczych wykonujących działalność leczniczą w rodzaju stacjonarne i całodobowe świadczenia zdrowotne
REKLAMA
Dziennik Ustaw
REKLAMA
REKLAMA